Dimérisation des récepteurs
Presque tous les types de récepteurs se dimérisent après fixation de leur ligand (à l'exception des récepteurs qui forment déjà de grands complexes protéiques tels que le récepteur nicotinique de l'acétylcholine). Souvent, le ligand possède deux ou plusieurs sites de liaison ce qui lui permet de regrouper plusieurs récepteurs (dimérisation ou oligomérisation). Pour d'autres c'est la fixation du ligand qui induit un changement de conformation du domaine extracellulaire révélant ainsi un site d'interaction entre deux récepteurs. Dans d'autres cas, enfin, c'est l'effecteur intracellulaire qui regroupe deux récepteurs. La dimérisation est nécessaire pour faire passer le signal vers l'intérieur (voir figure 13).
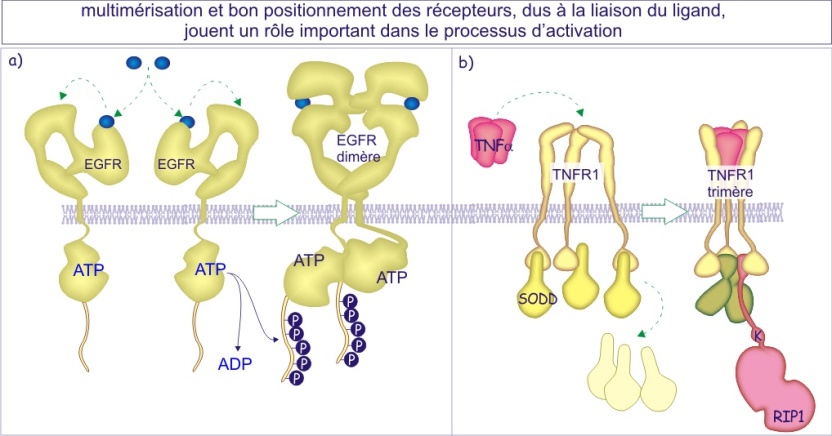
La raison d'être de la dimérisation n'est pas encore bien comprise, mais il semble qu'elle permette d'améliorer le contraste du signal (rapport signal sur bruit fond). En d'autres termes, elle diminuerait la probabilité de déclenchement d'un signal accidentel.
Remarque : Découverte de la dimérisation fonctionnelle des récepteurs
Dès la découverte des récepteurs de facteurs de croissance (EGFR et PDGFR), la nécessité de leur dimérisation est apparue évidente. En ce qui concerne les récepteurs de neurotransmetteurs des indices probants sur leur arrangement en oligomères (ce qui signifie qu'ils s'assemblent en deux (dimère) ou quelques unités (trimère, tétramère, pentamère etc) ont été notés dans les années 80. La démonstration de l'existence d'oligomères a été apportée à propos des récepteurs métabotropiques GABAB2 qui sont incapables de transmettre un signal à moins que les deux sous-types de récepteurs (les variants d'épissage 1 et 2) ne soient présents simultanément. Il est vraisemblable que l'hétérodimère GABAB1 - GABAB2 existe en tant qu'entité stable non dépendante de son état d'activation (liaison avec le ligand). En revanche, pour d'autres, tels que le récepteur glutamatergique (mGluR5), le récepteur V2 de la vasopressine, le récepteur de l'adénosine, le récepteur \(\delta\)-opioïde ou le récepteur cholinergique muscarinique, l'équilibre entre les formes monomérique et dimérique pourrait refléter leur état d'activation.
![]() Pour en savoir plus :
Pour en savoir plus :
« EGF dimérisation Schlessinger [pdf] » (4,3 Mo).
« GABA heterodimerization Bettler [pdf] » (167 ko).
Complément : Excursion 3 : Interaction entre récepteur et ligand et activation du récepteur: agonistes, agonistes-inverses et antagonistes.
Interaction entre récepteur et ligand et activation du récepteur: agonistes, agonistes inverses et antagonistes
On sait que les canaux ioniques sont des protéines qui se présentent sous différents états, typiquement “ouvert” ou “fermé”. Dans le cas des canaux activés par un ligand (par exemple le récepteur nicotinique de l'acétylcholine), la probabilité d'ouverture est considérablement augmentée lorsque le ligand est fixé.
Un équilibre entre les deux états existe également pour les récepteurs « non-canaux », un phénomène particulièrement bien étudié pour les récepteurs couplés aux protéines-G (GPCR ou récepteurs sept fois transmembranaires). La conception classique postulait que l'activation du récepteur exige la fixation effective d'un ligand (agoniste), qui induit un changement de la conformation, permettant ainsi l'interaction avec un effecteur (protéine G par exemple). Cependant, nous savons maintenant que le récepteur peut se trouver dans son état « actif » en absence de ligand. Ceci est illustré par les deux exemples suivants :
la surexpression du récepteur \(\beta\)2-adrénergique dans les cellules Sf9 d'un insecte, qui ne répondent normalement pas à l'adrénaline, entraîne la production d'AMPc (conséquence de l'activation de l'adénylate cyclase) sans qu'il y ait présence d'adrénaline.
une surexpression 200 fois supérieure à la normale du même récepteur\(\beta\)2-adrénergique du cœur d'une souris transgénique, est suffisante pour maximaliser la fonction cardiaque en absence d'adrénaline. Ceci peut être compris si le récepteur existe sous deux états conformationnels, l'un d'entre eux pouvant communiquer avec les effecteurs (R*). L'équilibre entre ces deux états existe en absence de ligand (voir figure E09).
Dans ce modèle, les agonistes conventionnels sont des molécules qui se lient aux récepteurs dans leur état actif (R*) et les y maintiennent, augmentant ainsi la proportion des récepteurs qui communiquent avec leurs effecteurs. Au contraire, il y a des « agonistes inverses » qui se fixent aux récepteurs dans leur état inactif et les y maintiennent, augmentant ainsi la proportion des récepteurs qui ne communiquent pas avec leurs effecteurs. Les agonistes augmentent le signal alors que les agonistes inverses le diminuent.
Lorsqu'il s'agit de récepteurs qui se dimérisent dans leur état actif (R*R*), par exemple les récepteurs \(\beta\)2-adrénergiques, il a été montré que l'addition de l'agoniste (adrénaline ou noradrénaline) favorise la formation de dimères, alors qu'au contraire, l'agoniste inverse (timolol) favorise l'état monomère des récepteurs.
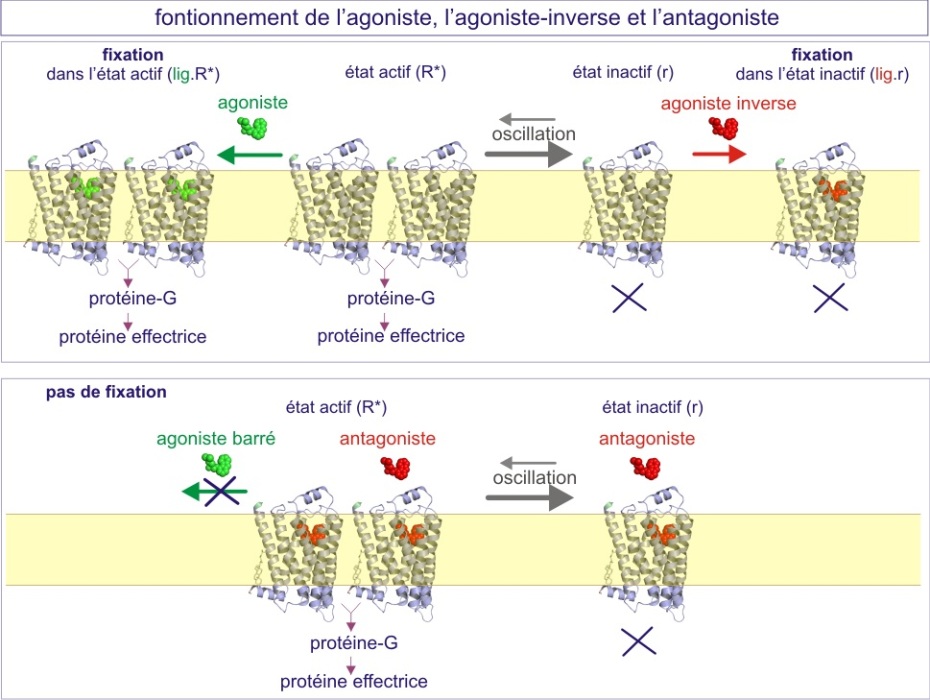
Les agonistes partiels sont définis comme des molécules qui se fixent aux récepteurs aussi bien dans leur conformation active qu'inactive, si bien qu'ils ne déclenchent qu'un signal peu contrasté. Ceci est favorable dans le cas du traitement de certaines déficiences cardiaques. Par exemple, un agoniste partiel \(\beta\)1-adrénergique (alprénolol ou oxprénolol) assurera un rythme cardiaque soutenu mais préviendra aussi l'excès de fréquence dû à une stimulation adrénergique (situation de stress).
Les antagonistes, enfin, sont définis comme des molécules qui se fixent aux récepteurs aussi bien dans leur conformation active qu'inactive sans déclencher de signal. Ils agissent en empêchant la fixation des agonistes. En général, les agonistes et antagonistes sont en compétition : augmenter la dose d'agoniste surmonte l'effet inhibiteur de l'antagoniste.
Des arguments montrent que la surexpression des récepteurs en soi, peut être responsable de certaines pathologies : par exemple, dans la schizophrénie on observe une surexpression (trois fois) du récepteur D4-dopaminergique dans le cortex frontal sans qu'il y ait une augmentation de dopamine (donc pas d'excès de ligand). L'administration d'un antagoniste qui ne diminue pas le nombre de récepteurs dans leur état actif (R*) et donc le signal « spontané », serait sans effet notable sur le signal du récepteur D4. En revanche, l'administration d'un agoniste inverse, en augmentant le ratio des récepteurs inactifs, aurait un effet bénéfique.