5.2.2.3 - Modèles d'évolution
Toutes les substitutions qui se produisent à l'échelle de l'évolution ne sont pas observables à travers un alignement de séquences. Lorsque les temps évolutifs sont longs, des substitutions anciennes peuvent être masquées par des substitutions plus récentes. Au delà des substitutions multiples d'autres événements telles que les substitutions parallèles[1] , convergentes, de réversion peuvent réduire / saturer une partie du signal évolutif.
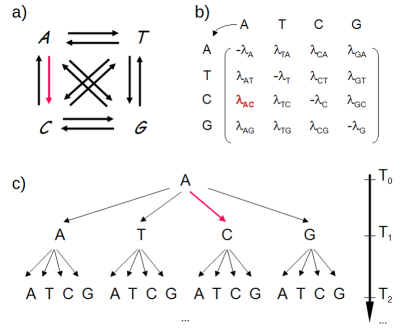
Pour répondre à ce problème des modèles markoviens de l'évolution des séquences ont été développés. Le principe général de ces modèles consiste à décrire l'ensemble des substitutions possible (fig. 5.35a) pour chaque unité de temps évolutif considérée (c). Les substitutions sont décrites à travers une matrice de substitution (b). Le nombre de substitution estimés pour un site sur un intervalle de temps T devient :
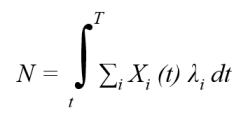
où Xi est la fréquence du nucléotide i dans la séquence et λi la probabilité pour le nucléotide de muter sur l'intervalle de temps dt.
A partir de cette équation générale, on peut dériver plusieurs équations permettant d'estimer :
le nombre théorique de substitutions accumulées sur une séquence pendant un intervalle de temps T
la distance théorique entre deux OTU (incluant les substitutions observables et non observables)
la probabilité d'avoir un nucléotide particulier après un intervalle de temps T
la probabilité π d'avoir une substitution[2] entre deux OTU dérivant d'un ancêtre commun après un intervalle de temps T
la probabilité théorique π peut être estimé à partir des données par le comptage des substitutions observées (distance observée p) dans les alignements de séquences. Ainsi, par le biais :
des équations générées par les modèles d'évolution
de « l'estimation de leur paramètres » à partir de la mesure de la divergence observée,
on peut déduire les distances évolutives corrigées des séquences considérées.
A partir de ce modèle d'évolution général, des modèles spécifiques ont été déclinés, qui varient par le traitement des différents types de substitutions et des fréquences des différents nucléotides dans les séquences (fig.5.36). Le modèle le plus simple et sans doute le plus connu est celui de Jukes et Cantor[3] qui considère que tous les types de substitutions ont la même probabilité de se produire et que la fréquence des nucléotides sont équilibrées (identiques). On peut également citer le modèle de Kimura à deux paramètres ( K2P[4]) qui dans la matrice de substitutions distingue les transitions des transversions, dont on sait qu'elles se produisent à des fréquences différentes dans les génomes.
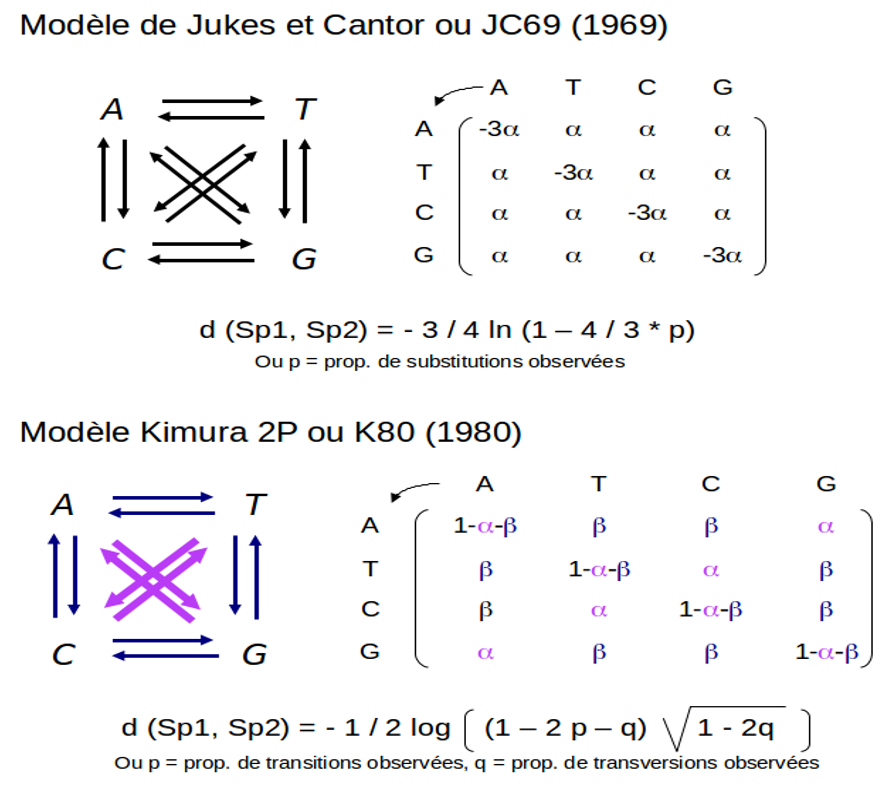
Il est à noter enfin que si les modèles d'évolution sont en théorie applicable tout autant sur les acides aminés que sur les nucléotides, la complexité des calculs que ces modèles sous-tendent lorsqu'il faut considérer 20 acides aminés (contre 4 nucléotides) fait que cette approche n'est pas utilisée dans l'estimation des distances évolutives sur des séquences de protéines. Dans ce cas, les matrices de substitution sont le plus souvent conçues à partir de l'analyse d'alignements protéiques de références (eg. BLOSUM, fig. 5.37)
Exemple : matrice BLOSUM
La matrice BLOSUM (BLOcks SUbstitution Matrix) (fig. 5.37) est calculée à partir de séquences de références bien conservées ("blocks" correspondant à des domaines de nombreuses familles protéiques). Chaque conservation et chaque substitution est analysée statistiquement et un score lui est attribué :
une valeur nulle équivaut à une substitution de fréquence moyenne
une valeur positive correspond à une conservation ou une substitution sur-représentée
une valeur négative correspond à une substitution rare et donc potentiellement plus défavorable
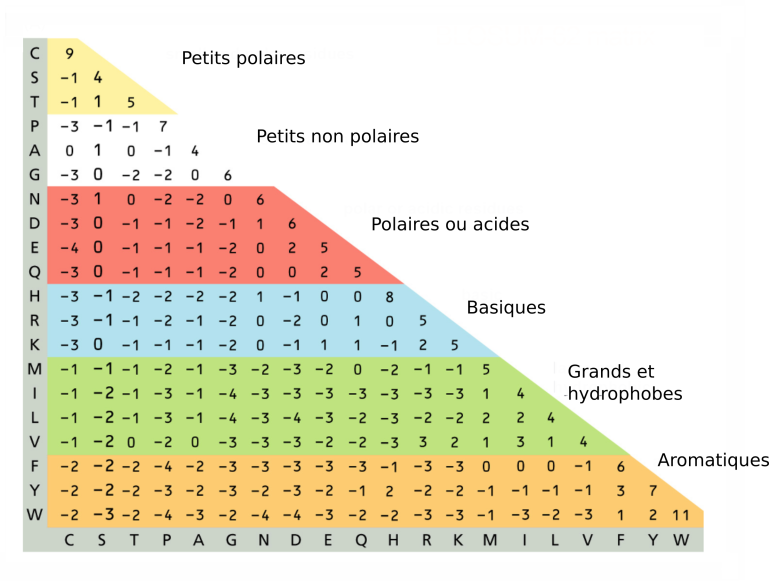
On remarque par exemple dans cette matrice que :
l'acide aminé tryptophane (W) est l'acide aminé le mieux conservé (W et W à la même position est la situation ayant le score le plus élevé : 11).
les substitutions F/Y ou V/I sont les plus fréquentes (score = 3)
les substitutions les moins fréquentes sont entre autre P/W, L/G, E/C etc. (score -4)
On remarque que ces dernières correspondent à des changements de famille des acides aminés qui ont la plus grande probabilité de modifier la fonction de la protéine.
Lors de la comparaison de protéines, orthologues, plus le score sera faible, plus la différence sera significative.