Remarque sur la fonction des peroxysomes
La fonction des peroxysomes est évoquée rapidement ici car nous n'y reviendrons pas ultérieurement.
Remarque : Découverte du peroxysome
Le peroxysome fut pour la première fois décrit en 1954 dans la thèse d'un étudiant suédois (J. Rhodin) comme un petit organite à simple membrane (microsome) présent dans les cellules du rein de souris. En 1966, cet organite était retrouvé, par microscopie électronique, dans une fraction microsomale (avec les mitochondries et les lysosomes) cellulaire par l'équipe de Christian de Duve. Par la suite, l'équipe a pu séparer les peroxysomes des autres constituants par centrifugation en gradient de densité, et cela grâce à leur remarquable haute densité.
Pour ses découvertes concernant l'organisation fonctionnelle et structurale de la cellule, Christian de Duve a reçu le prix Nobel « Physiologie et Médecine » en 1974 (partagé avec George E. Palade et Albert Claude).
Leur fonction métabolique majeure est la \(\beta\)–oxydation de longues et très longues chaînes d'acides gras (plus que 18 carbones) qui, en aboutissant à la formation de chaînes plus courtes, alimentera les processus mitochondriaux de \(\beta\)–oxydation. Ils doivent leur nom au fait que la première étape d'oxydation des acides gras se traduit par la formation de peroxyde d'hydrogène (H2O2) (voir figure 6 ).
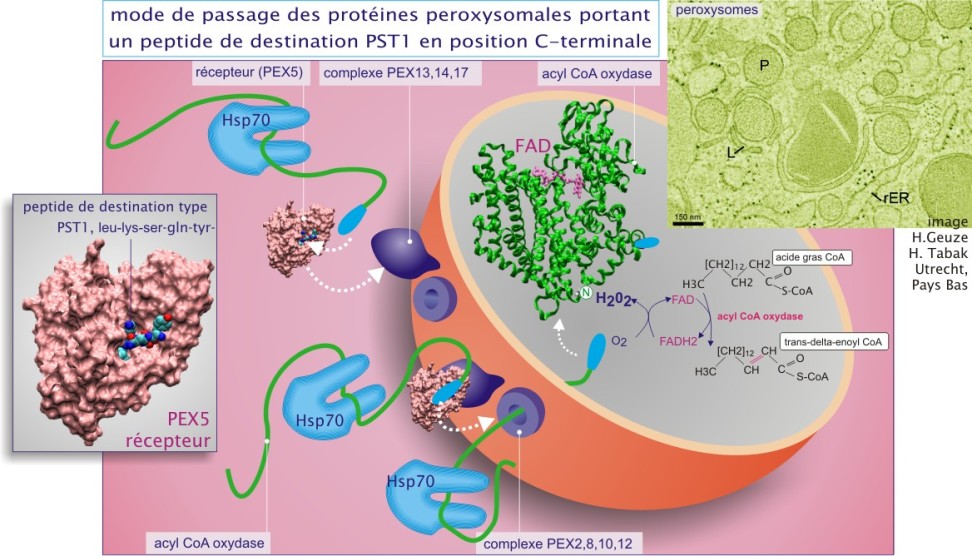
pour plus de détail sur la beta-oxydation peroxysomale.
En dehors de la \(\beta\)–oxydation des longues chaînes d'acides gras, le peroxysome est aussi impliqué dans la synthèse de la bile, du cholestérol et du plasmalogène (etherlipide) et également dans le métabolisme des acides aminés et des purines.
La place du peroxysome dans le métabolisme cellulaire est illustrée par l'existence de nombreuses pathologies associées à son dysfonctionnement, lui même dû à des défauts génétiques :
mutation d'une des protéines impliquées dans le transport au travers de la membrane (les protéines PEX, voir le paragraphe « Transport des enzymes peroxysomales matricielles à travers la membrane »),
mutation d'une des protéines de la matrice peroxysomale.
Complément : Excursion : le métabolisme lipidique du peroxysome
Différences entre \(\beta\) –oxydation mitochondriale et peroxysomale
Deux différences majeures existent chez les mammifères :
les mitochondries oxydent des chaînes moyennes à courtes d'acides gras alors que les peroxysomes oxydent de longues et même très longues chaînes (plus de 18 carbones) ;
les mitochondries utilisent une acyl–CoA–déshydrogénase pour convertir l'acyl–CoA en énoyl–CoA, une enzyme qui transfère les électrons sur le FAD (formant ainsi FADH2) et donc sur la chaîne respiratoire. En revanche, les peroxysomes utilisent une acyl–CoA–oxydase qui transfère les électrons sur l'oxygène, produisant ainsi du peroxyde d'hydrogène, détruit ensuite par la catalase. Le reste de la voie de \(\beta\)–oxydation est identique sauf que les enzymes peroxysomales et mitochondriales sont des entités protéiques distinctes car codées par des gènes distincts (différant au moins par les messages codant des peptides de destination différents).
Pathologies
Une première catégorie de désordres est le résultat d'un défaut dans une seule enzyme peroxysomale. Ces désordres, parmi toute une liste, incluent l'hyperoxalurie type I (alanine:glyoxylate aminotransférase), la maladie de Refsum (manque de phytanoyl–CoA hydroxylase), l'adrénoleucodystrophie liée au chromosome–X (ALDP), etc.
Une seconde catégorie de désordres résulte d'une déficience dans la biogenèse du peroxysome qui affecte toutes ses voies métaboliques, conséquence d'une mutation dans l'un des gènes de PEX (récepteurs et transporteurs de protéines matricielles). Ces désordres sont connus sous l'appellation PBD, pour peroxisome biogenesis disorders.
Quelques exemples : le syndrome de Zellweger (ZS), l'adrénoleucodystrophie néonatale (NALD), la maladie de Refsum infantile (IRD) et la chondrodysplasie punctata rhizomélique type I (RCDP).
Présentation clinique
Les caractéristiques cliniques sont une hypotonie (manque de tonus), un dysmorphisme craniofacial, un retard mental sévère, des défauts sensoriels, une faiblesse musculaire, une hypomyélinisation et une hépatomégalie. Pour la plupart, les patients meurent de ces désordres dans leurs deux premières années.