2. Le docking
Définition :
Le docking (ou "amarrage moléculaire") est une approche numérique de modélisation moléculaire. Elle cherche à identifier la meilleure orientation entre 2 molécules sur la base de leur coordonnées dans des états pris séparément.
Le principe du docking repose sur 3 étapes :
Représentation des systèmes moléculaires
Établissement d'un algorithme de recherche des interactions
Établissement d'une fonction permettant d'évaluer l'interaction entre les partenaires
Les systèmes moléculaires sont représentés majoritairement au niveau atomique. Chaque atome de la molécule considérée est représenté de manière explicite. Il s'agit généralement d'une sphère, chargée électriquement, qui va interagir avec ses voisins.
On se rapportera au cours de modélisation moléculaire (mécanique moléculaire) pour une version détaillée :
http://uel.unisciel.fr/chimie/modelisation/modelisation_ch02/co/modelisation_ch02.html
Avec cette approche, on peut comparer les énergies des différentes conformations d'un système moléculaire.
Toutefois, le nombre de conformations est très grand ! Les dénombrer en totalité est très coûteux en temps de calcul :
Le nombre de conformères N peut être estimé par la formule :
\(N = T^{\frac{360}{i}}\) où T est le nombre de liaison qui peuvent tourner, i est le degré avec lequel on fait tourner une liaison.
Exemple :
Pour 10 liaisons dans une molécule flexible qu'on envisage de faire tourner par pas de 30°, on dénombre 1012 conformations possibles !
Si on passe à une molécule de 20 liaisons avec un incrément plus précis de 10°, on trouve 7.1046 conformère pour lesquels il faut estimer l'énergie afin de les classer et trouver la plus stable.
Il existe différentes façons de faire des recherches dans cet espace très complexe. La plus simple (recherche naïve) est de générer tous les conformères et à chaque fois de calculer l'énergie par une fonction de mécanique moléculaire. Mais ce type de calcul est très chronophage.
Méthode :
La méthode Monte-Carlo permet de trouver les conformères les plus stable en un temps plus court.. Elle est détaillée dans ce chapitre :
http://uel.unisciel.fr/chimie/modelisation/modelisation_ch04/co/modelisation_ch04.html
Méthode :
D'autres approches, comme les algorithmes génétiques permettent aussi de gagner du temps. Sans entrer dans le détail, il s'agit de coder chaque variable structurale d'une molécule dans un équivalent de chromosome. Chaque incrément d'angle ou de distance est associé à une position dans le chromosome.
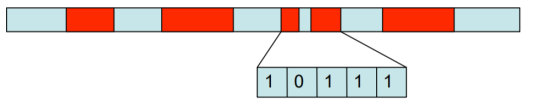
On commence alors par générer une population de génomes. Chaque chromosome est associé à une énergie de conformation. Puis on applique des croisements ou des mutations pour changer les structures. Comme dans la biologie de l'évolution, les meilleurs survivront ! Ici les meilleurs candidats sont ceux avec les énergies de conformation les plus stables. À la faveur de croisement, on va sélectionner les structures les plus stables.
À l'issue de la mise en œuvre des ces algorithmes de recherche, on dispose donc de structures de molécules ou de récepteurs dans des conformations stables.
Leur énergie d'interaction peut être calculée en utilisant la mécanique moléculaire, qui prend en compte des termes représentant les interactions du chapitre précédent.
On classe alors les structures ligand/récepteur (appelées “poses”) par ordre de stabilité avec l'idée que les poses les plus stables seront celles potentiellement observées expérimentalement.
Remarque :
La qualité de la fonction dite “de score” (basée sur l'énergie d'interaction la plupart du temps) n'est pas nécessairement représentative de la valeur expérimentale. Les critères demandés à la fonction de score sont :
Donner le meilleur score à la structure expérimentale qu'elle est censée prédire
Donner un bon classement relatif parmi une série de ligands candidats
Être calculée rapidement pour pouvoir cribler des bases de données de grande taille
Simulation :
Interactions dans des cavités