TP 2 – Docking de différents agonistes du récepteur ß-2 adrénergique.
Partie
Objectif : Évaluation d'un protocole de docking pour retrouver le site d'interaction du carazolol dans le récepteur ß-2 adrénergique.
Avant-propos :
Le logiciel utilisé dans ce TP, UCSF Chimera, est un logiciel de visualisation et servira à analyser les résultats. UCSF Chimera est multi-plateforme et gratuit pour un usage non commercial. Il n'est pas possible de lancer des simulations de docking avec ce logiciel, en revanche, il servira d'interface pour lancer Autodock Vina sur le serveur proposé par UCSF Chimera ou directement sur un ordinateur (une installation préalable du logiciel de docking sera nécessaire). Avant de vous lancer dans ce TP, il faudra installer le logiciel UCSF Chimera et se familiariser avec l'interface. Pour cela, le site de l'éditeur, en anglais, est très bien illustré : https://www.cgl.ucsf.edu/chimera/
Fichiers utiles pour le TP :
Question
Partie 1/ Préparation des simulations de docking
Ouvrir avec UCSF Chimera les structures PDB du récepteur ( 2RH1_rec.pdb) puis du ligand ( 2RH1_lig.pdb). Les structures proviennent de la structure cristallographique 2RH1 et sont également téléchargeables sur le site http://www.rcsb.org
Ajouter les atomes d'hydrogène manquant sur la structure du récepteur (et uniquement du récepteur ! Ceux du ligand sont déjà présents dans le fichier fourni) et attribuer les champs de forces
AMBER ff03.r01etGaistegerrespectivement au récepteur et au ligand.Aide : Pour ajouter les atomes d'hydrogène, ouvrir le menu
Tools > Structure Editing > AddH. Pour sélectionner un champs de force, utiliser le menuTools > Structure Editing > Add charges.Préparer les paramètres de simulations de docking en ouvrant le menu
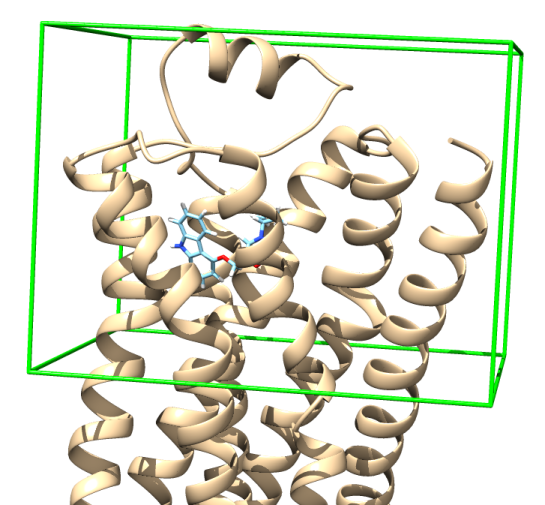
Tools > Surface/Binding Analysis > Autodock Vina. Dans la fenêtre qui s'ouvre : choisir un nom de fichier de sortie (par exempledock_carazolol), sélectionner le récepteur (2RH1_rec.pdb) et le ligand (2RH1_lig.pdb). Définir l'espace de recherche (taille et position du centre de la grille). Nous vous conseillons les paramètres suivants :Paramètres pour le docking avec AutoDock Vina Center
-36.381
4.757
6.904
Size
41
31
34
Dans le sous menu
Receptor options, sélectionner dans l'ordre les choixfalse-true-true-true-true-false. Dans le sous menuLigand options, sélectionner dans l'ordre les choixfalse-true.Enfin, dans le sous menu
Executable locationchoisir le serveur opal puis lancer le calcul. Dans le cas d'une installation locale du logiciel Autodock Vina, sélectionner le répertoire contenant l’exécutable.
Question 1 :
Pourquoi est-il nécessaire d'ajouter les atomes d'hydrogène manquant ?
Solution détaillée
Une structure cristallographique ne contient pas d'atome d'hydrogène. En mécanique classique, les champs de force sont basés sur une description des interactions par paires d'atomes. Dans la plupart des cas, il est donc nécessaire de traiter explicitement tous les atomes du système moléculaire. En revanche, dans certains champs de force, il est possible de s'affranchir des interactions n'apportant pas de spécificité au système moléculaire. Par exemple, dans les champs de force ‘united-atom' les atomes d'hydrogène d'un système aliphatique sont regroupés avec les atomes de carbone auxquels ils sont liés. Le rayon de van der Waals de l'atome de carbone ainsi que sa charge atomique sont ainsi modifiés pour prendre en compte non pas un seul atome mais un « grain » plus gros. C'est également la philosophie des champs de force que l'on appelle « gros-grains » où un ensemble d'atomes sont regroupés pour former des « pseudo-atomes » (ou groupe d'atomes). Cela permet de simplifier les calculs en diminuant le nombre d'interactions à calculer.
Question
Question 2 :
Quelles sont les limitations inhérentes à une simulation de docking ?
Solution détaillée
Il faut être conscient des limites de la méthode de calcul. Tout d'abord, la fonction de score est généralement basé sur une approche empirique pour estimer l'énergie d'interaction. Certains termes énergétiques (comme le terme entropique) sont très difficiles à estimer. Le score de docking doit donc être considéré avec une certaine prudence. Un écart de moins d'1 kcal/mol n'est probablement pas significatif.
Ensuite la définition de l'espace de recherche limite forcément les solutions qui peuvent être obtenues. Un espace de recherche trop petit ne permettra pas d’échantillonner des solutions de docking alternatives. A l'inverse, un espace de recherche trop large risque de générer des faux positifs dans les solutions de docking. L'idéal est de limiter l'espace de recherche aux régions connues (d'après les données expérimentales) pour contenir le site de liaison.
Enfin, il existe plusieurs niveaux de complexité dans une simulation de docking. Tout d'abord le ligand et le récepteur peuvent être considérés comme des blocs rigides. La simulation de docking revient à résoudre un problème d'optimisation à 6 dimensions (3 dimensions pour la rotation et 3 pour la translation). C'est le cas le plus simple.
Il est possible de donner de la flexibilité au ligand en autorisant les modifications des angles dièdres (également appelées rotors) non contraints. Ce sont en effet les degrés de liberté les plus important lors de l’échantillonnage de l'espace conformationnel d'une molécule.
Enfin, il est possible d'ajouter de la flexibilité au récepteur. Plusieurs approches existent. Il est par exemple possible d'autoriser certains rotors des chaînes latérales des résidus de la poche de liaison à être modifiés. Cela permet au récepteur de s'adapter à la présence du ligand et de former des interactions spécifiques au cours de la simulation. il est également possible de modifier l'ensemble de la structure du récepteur en suivant les modes normaux de plus basse fréquence qui auraient pu être pré-calculés. Ces mouvements moléculaires sont censés représenter les mouvements de plus grande amplitude susceptibles d'intervenir lors de l'association ligand-récepteur.
Question
Partie 2/ Analyse des résultats de docking
Question 3 :
Après quelques minutes, un menu s'ouvre avec les différentes solutions de docking proposées par le logiciel Autodock Vina. Inspecter les différentes solutions. Est-ce cohérent avec la pose cristallographique ?
Solution détaillée
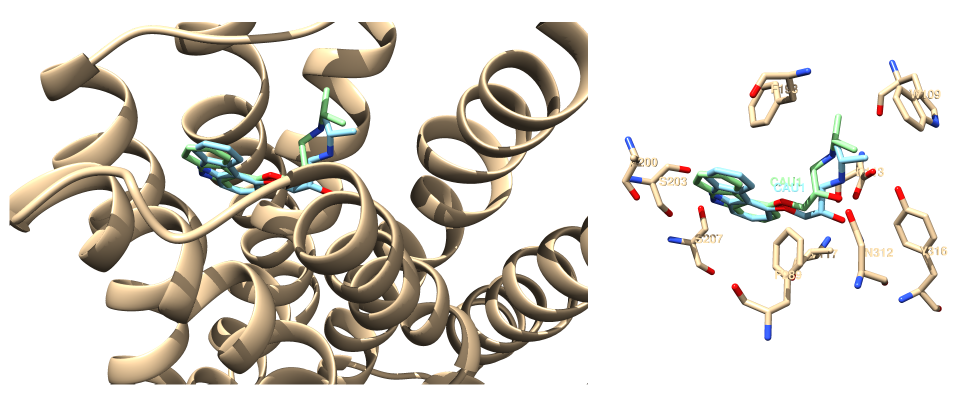
Il semble que la solution classée 1 d'après la fonction de score Vina positionne correctement le ligand dans la cavité de liaison. La figure 2 montre la superposition de la structure expérimentale du ligand (à gauche) avec celle prédite par docking et fait ressortir les mêmes d'interactions ligand-récepteur (à droite).
Question
Question 4 :
Calculer le RMSD entre les différentes solutions générées par le docking et la référence (pose cristallographique).
Résumer les résultats dans un tableau comportant le RMSD et le score d'interaction.
Analyser les résultats et conclure sur le protocole.
Aide méthodologique
Pour cela il faudra ouvrir le mode ligne de commande (Menu Favorites > Command Line) et taper la commande suivante :
rmsd #1@C?,C??,N?,N??,O?,O?? #3.1@C?,C??,N?,N??,O?,O??
Remarque: Cette commande permet de calculer le RMSD entre tous les atomes lourds de la position RX et de la première solution du docking.
Voici quelques explications pour modifier la commande :
#1: devrait correspondre au modèle RX du ligand (#2 devrait être le modèle RX du récepteur)#3.1: devrait correspondre au 1er modèle généré par docking (#3.2 au 2e, #3.3 au 3e, etc.)@: les lettres qui suivent ce symbole précise sur quels atomes le RMSD est calculé. Ici tous les atomes dont les noms commencent par C, N et O.
Solution détaillée
Le tableau ci-dessous résume les cinq premières solutions de docking classées à l'aide de la fonction de score Autodock Vina. On remarque que la meilleure pose de docking est ici effectivement celle qui se rapproche le plus de la structure expérimentale (c-a-d celle dont le RMSD du ligand est le plus petit). Dans ce cas, on peut considérer que l'algorithme de docking réussit à discriminer la bonne solution parmi toutes celles générées.
modèle | RMSD ligand (Å) | Score Vina (kcal/mol) |
#3.1 | 1.1 | -9.5 |
#3.2 | 5.8 | -8.8 |
#3.3 | 6.3 | -8.8 |
#3.4 | 3.6 | -8.7 |
#3.5 | 10.1 | -8.1 |