6.4 Repliement des protéines
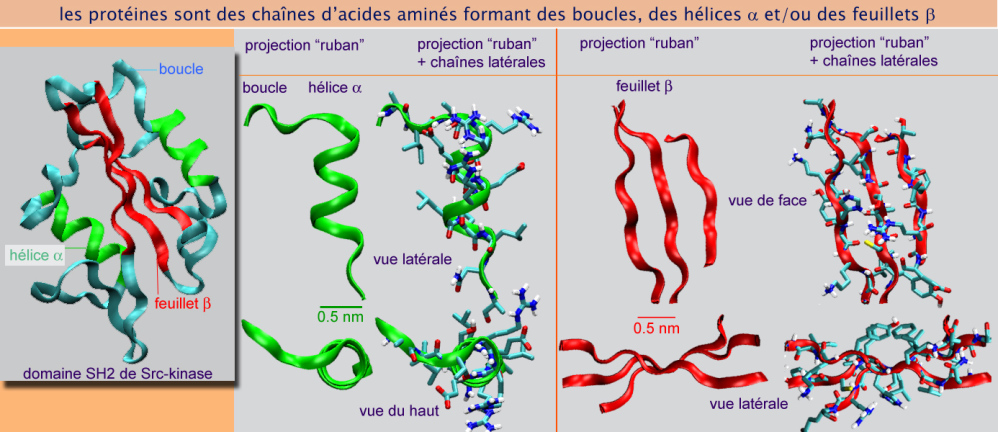
Une protéine est une chaîne polypeptidique dont le repliement fait apparaître un ou plusieurs domaines, chacun d'entre eux étant constitué d'hélices \(\alpha\), de feuillets \(\beta\) et de boucles (voir figure 18). Le processus par lequel cette chaîne acquiert une forme tridimensionnelle correcte de façon à assurer sa fonction biologique, est appelé repliement protéique. Bien que certaines chaînes polypeptidiques se replient spontanément (75%), d'autres requièrent l'assistance de protéines qualifiées de chaperonnes. De nombreuses chaperonnes portent le nom de heat-shock protein (Hsp), protéines de choc thermique, car fortement exprimées lors de températures élevées (jusqu'à 430 C). Les chaperonnes ont des formes variées mais ont en commun la capacité d'héberger des séquences hydrophobes (séquences riches en glycine, alanine, valine, leucine, thréonine ou isoleucine). Elles « offrent » un refuge temporaire à certaines parties des protéines naissantes. Une fois la chaîne peptidique en formation suffisamment longue pour cacher les sites hydrophobes à l'intérieur de sa structure secondaire, la chaperonne la libère après hydrolyse de l'ATP. La nouvelle protéine peut alors se replier par elle-même.
Le repliement peut être également déterminé par l'établissement de ponts disulfures entre deux résidus cystéine éloignés l'un de l'autre sur la chaîne protéique. Cette opération ne s'effectue pas dans le cytoplasme mais uniquement dans le réticulum par lequel transitent les protéines membranaires et celles qui sont destinées à l'exportation (exocytose). Ces protéines subissent généralement aussi un processus de glycosylation (maturation protéique) qui sera détaillé dans une autre ressource.
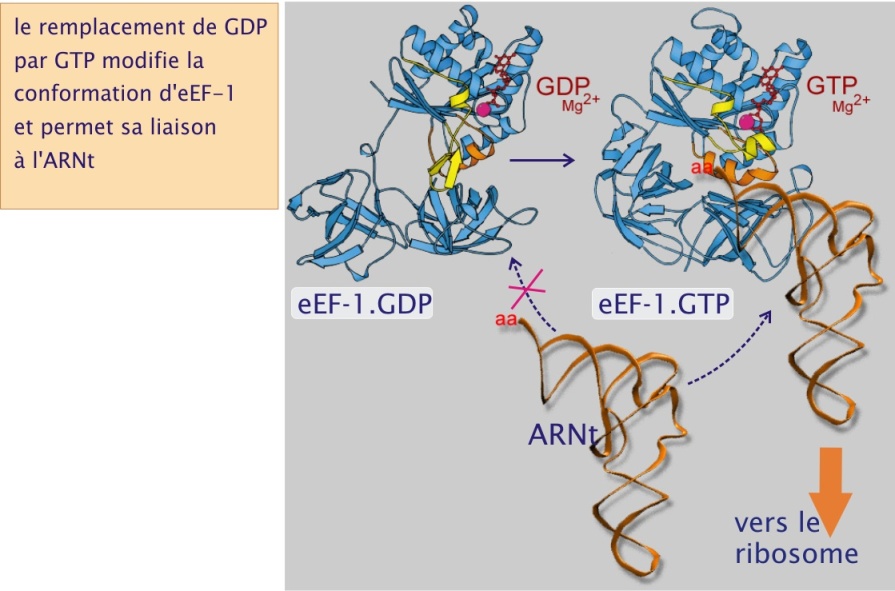
La raison d'être du repliement est la recherche d'une relative stabilité moléculaire, compromis entre rigidité structurale et fonction biologique qui nécessite souvent de petites modifications de conformation (d'une amplitude de l'ordre du nanomètre). Ceci est particulièrement vrai pour les protéines globulaires dont des exemples ont été évoqués précédemment, comme les pompes membranaires, les enzymes et les éléments du cytosquelette, actine, myosine et tubuline. Pour ces protéines, la fixation covalente de phosphate ou la liaison de nucléotides triphosphate ( ATP ou GTP) et leur hydrolyse subséquente, détermine leur conformation et par conséquent leur fonction (transport des ions, réactions chimiques, mouvement, polymérisation ou dépolymérisation). En matière de synthèse protéique, un bon exemple de repliement flexible est donné par le facteur d'élongation eEF–1. Ce facteur s'attache à l' ARNt lorsque il est lié au GTP (voir figure 19). Quand la liaison codon - anticodon réussit à l'intérieur du ribosome le GTP est hydrolysé (en GDP et Pi). Il s'ensuit un changement conformationnel qui détache le facteur eEF–1 et rend ainsi accessible le dernier acide aminé sur lequel sera transférée la chaîne polypeptidique en formation (en provenance de l' ARNt fixé sur le site P) (voir figure 19).
Complément : Excursion : repliement des protéines
Les premiers indices sur le phénomène de repliement qui confère aux protéines une structure dite secondaire, ont été apportés, aux environs de 1951, par les travaux de Linus Pauling et Robert Corey (California Institute of Technology, Pasadena, USA). Ensemble, ils découvrirent que fondamentalement, les structures secondaires protéiques étaient composées d'éléments nommés hélice \(\alpha\) et feuillet \(\beta\) (voir figure 30). De leurs travaux théoriques ils prédirent que ces structures étaient énergiquement favorables (et donc stables).
Pour plus d'information :

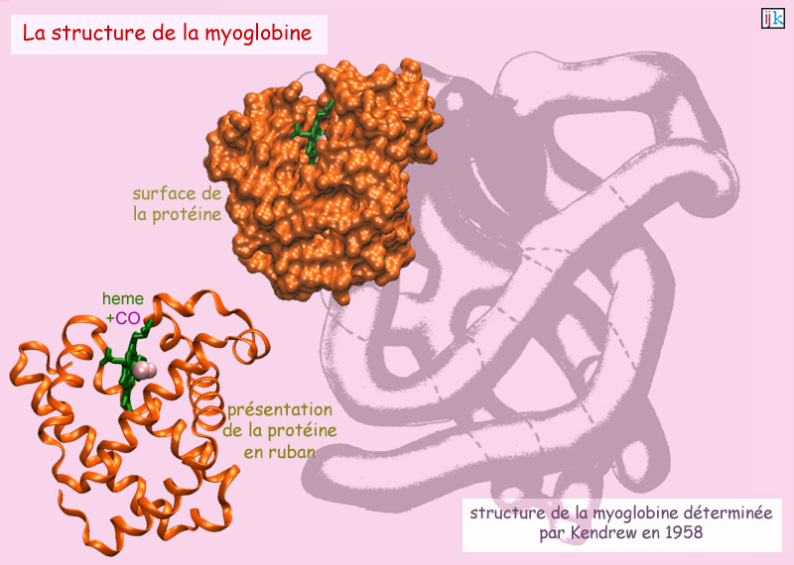
Plus tard, en 1953, quand Watson et Crick découvrirent la structure de l'ADN en double hélice régulière et symétrique, on admit implicitement que les protéines aussi était agencées en structures hautement symétriques formées d'hélices \(\alpha\) et/ou de feuillets \(\beta\). Si bien que, lorsqu'en 1958, John Kendrew (Medical Research Unit au Cavendish Laboratory, Cambridge, Royaume Uni) obtint la première image rayon X de la structure cristallographique de la myoglobine (protéine globulaire de 150 acides aminés), il fut désappointé de constater le repliement très irrégulier de la protéine : en accord avec la théorie de Pauling, elle comportait bien des hélices \(\alpha\), mais qui étaient arrangées de façon apparemment désordonnée (voir figure 31).
Selon John Kendrew : « Perhaps the most remarkable features of the molecule are its complexity and its lack of symmetry. The arrangement seems to be almost totally lacking in the kind of regularities which one instinctively anticipates, and it is more complicated than has been predicted by any theory of protein structure.»
Au début des années 70, Christian Anfinsen (Laboratory of Cellular Physiology and Metabolism du National Heart Institute of the National Institutes of Health, Bethesda, USA) montra que la structure tertiaire d'une protéine (la ribonucléase pancréatique bovine) est déterminée seulement par sa séquence primaire en acides aminés. Si la protéine se déplie artificiellement, elle revient à son état initial par elle-même, sans intervention de « replieur » ou de « sculpteur », phénomène expliqué par la thermodynamique de la conformation spatiale des protéines.
Selon Anfinsen : « ... the molecular structure of a native protein in its physiological environment is the one in which the Gibbs free energy of the system is the lowest » (Nobel speech, 1972).
Ceci est vrai pour de nombreuses protéines mais pour d'autres, il y a nécessité d'intervention de protéines « replieuses et sculpteuses », que l'on a dénommées chaperonnes. A ce jour, après avoir élucidé la structure de milliers de protéines, on ne sait toujours pas vraiment comment une séquence d'acides aminés de départ trouve sa conformation spatiale définitive. Par exemple, nous ne pouvons pas expliquer pourquoi et comment la chaîne polypeptidique de l'hémoglobine forme sept hélices \(\alpha\) reliées entre elles par des boucles et pourquoi le transporteur de la membrane interne de la mitochondrie forme un tonneau \(\beta\) type clé Grecque et des hélices \(\alpha\) comprimées.
L'importance des ces études sur la conformation spatiale des protéines est reflétée par l'attribution de plusieurs prix Nobel : à Linus Pauling en 1954 (chimie, pour sa recherche sur la nature de la liaison chimique et son application à l'élucidation de la structure de substances complexes), à John Kendrew en 1962 (chimie, pour ses études de la structure des protéines globulaires, prix partagé avec Max Perutz pour des études similaires sur l'hémoglobine) et à Christian Anfinsen en 1972 (chimie, pour son travail sur la ribonucléase, en particulier concernant la relation entre la séquence en acides aminés et la conformation biologiquement active, prix partagé avec Stanford Moore et William Stein).
Complément : Excursion : pathologies dues au repliement défectueux des protéines
Quand on fait cuire un oeuf, l'ensemble des protéines se solidifie en une masse insoluble et ne revient pas à l'état initial quand on le refroidit. Cela est dû à un repliement défectueux et irréversible des protéines (essentiellement l'albumine), nommé dénaturation. Au début du 20ème siècle, les pathologistes remarquèrent que certaines affections cérébrales étaient caractérisées par la présence de dépôts protéiques envahissant plusieurs structures nerveuses. C'est Alois Alzheimer aux environs de 1906 (dans le laboratoire d'anatomie de la clinique psychiatrique du Dr Kraepelin à Munich, Allemagne) qui remarqua la présence de « faisceaux hélicoïdaux de neurofibrilles » et de « plaques séniles » dans le cortex hippocampique de patients qui souffraient d'une démence sénile (actuellement connue sous le nom de maladie d'Alzheimer). Ces dépôts sont formés par des agrégats de protéines prématures, c'est-à-dire survenant rapidement (dans un intervalle de 0,1 à quelques secondes) avant que la protéine n'acquiert un repliement correct (qui évite toute agrégation). Il semble que l'accumulation de faisceaux hélicoïdaux de neurofibrilles se traduise par la mort neuronale (en particulier des neurones cholinergiques de la voie allant du striatum ventral à l'hippocampe).
Dans des maladies héréditaires rares nommées polyneuropathies amyloïdiques familiales (FAP) les nerfs périphériques et certains organes sont endommagés par des dépôts protéiques amyloïdes. Les études génétiques ont montré que l'anomalie est due à une mutation d'un seul acide aminé de la protéine transthyrétine (transporteur du rétinol et de la thyroxine (T3)). La protéine mutée est moins stable que la molécule origine dans des conditions d'acidité modérée. Les conséquences de cette mutation se manifestent lors des processus de destruction, pendant lesquels cette protéine se déplie pour entrer dans le protéasome : elle forme alors des agrégats moléculaires et, par conséquent, des dépôts destructeurs.