Phosphorylation due aux protéines kinases
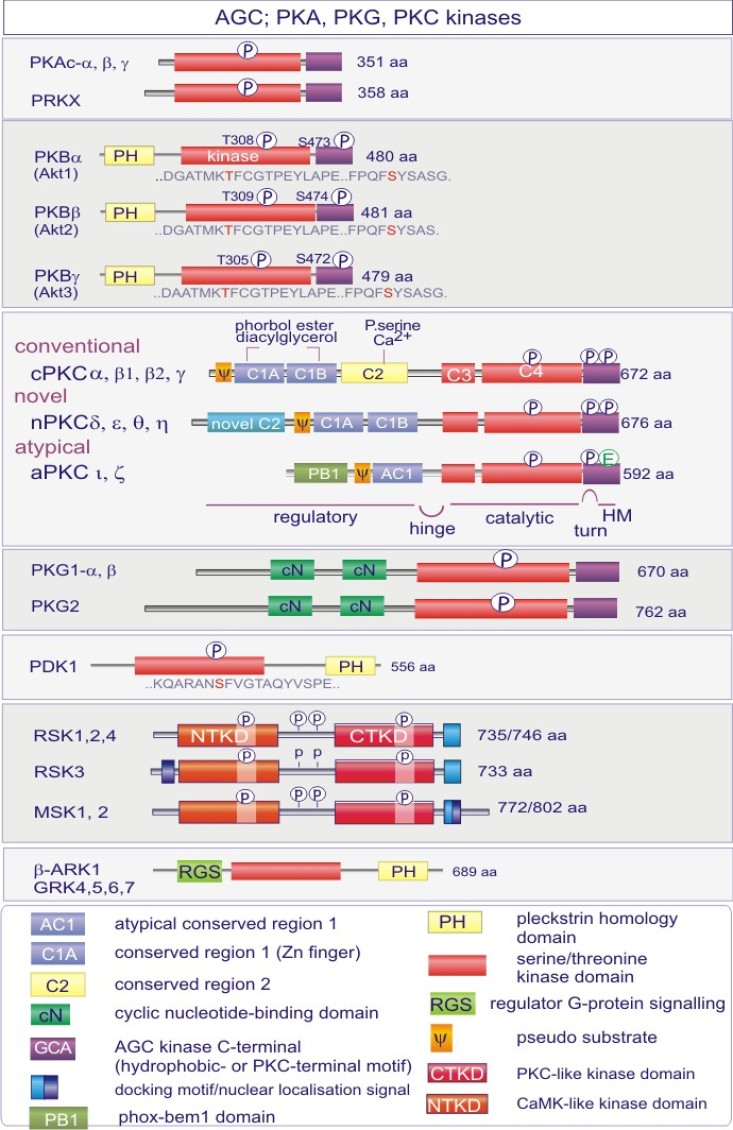
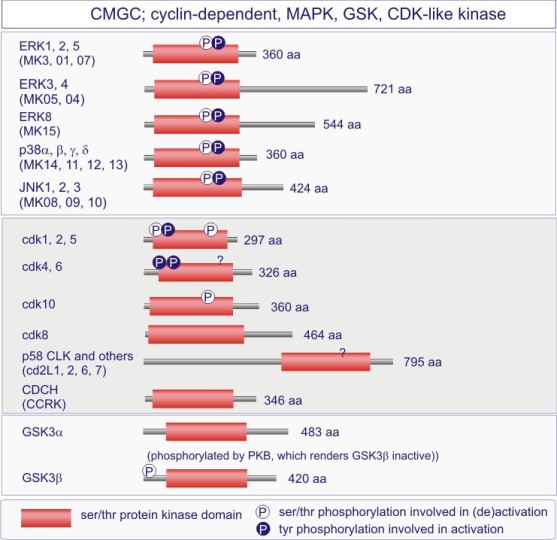
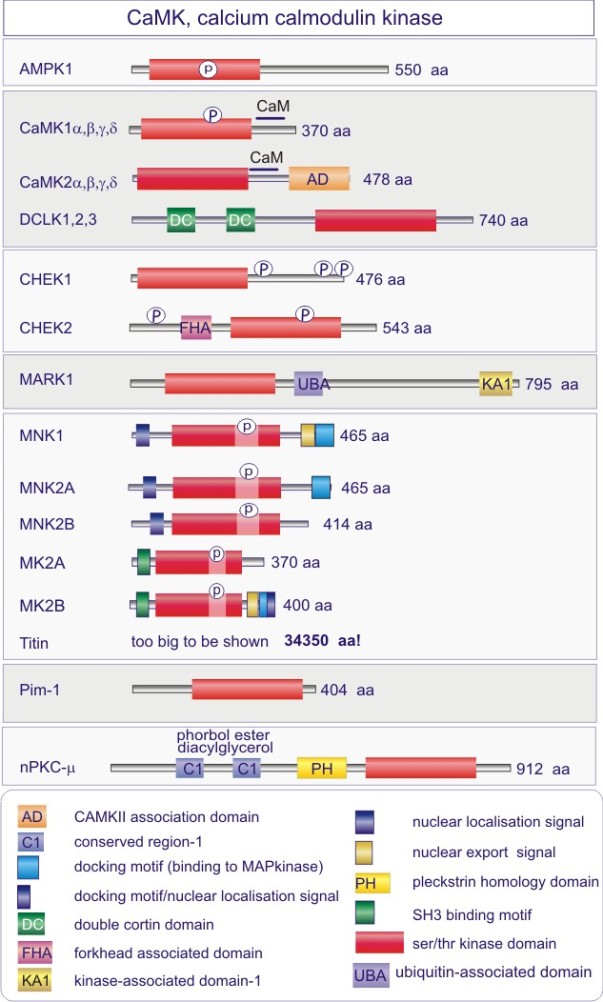
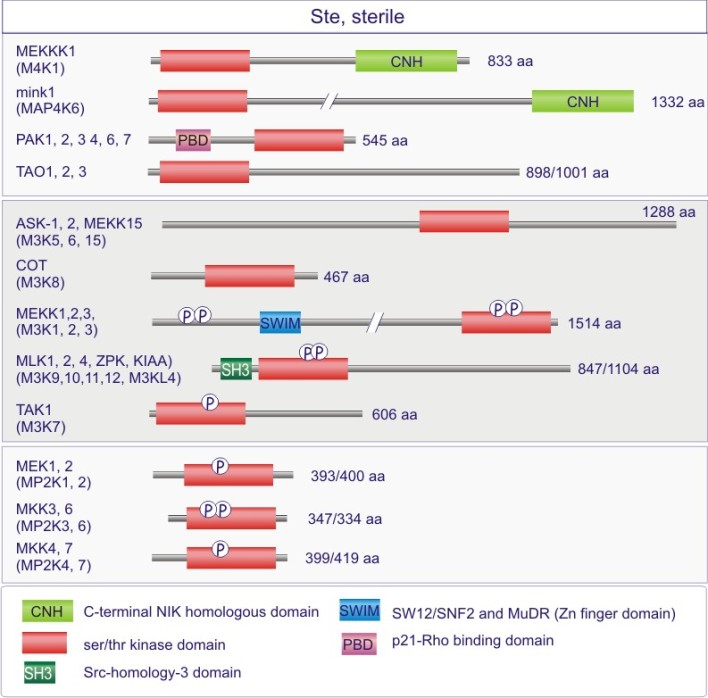
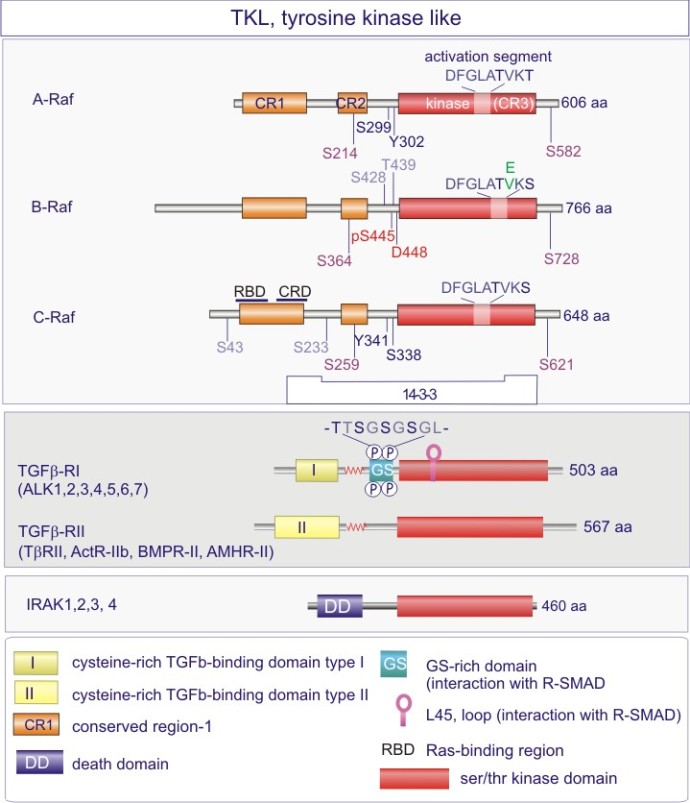
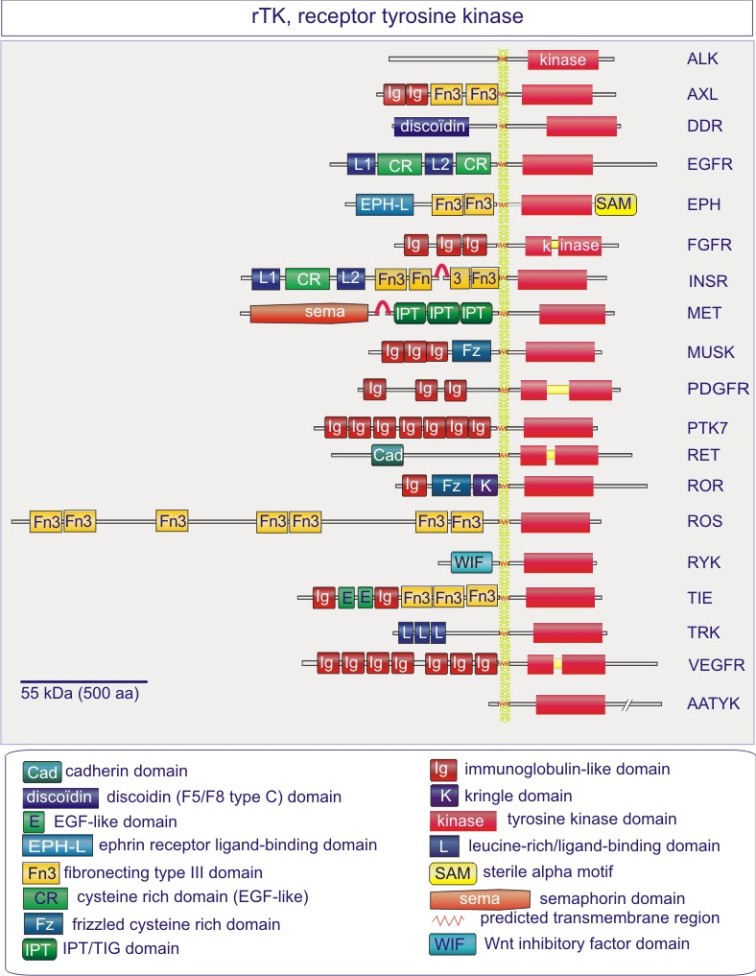
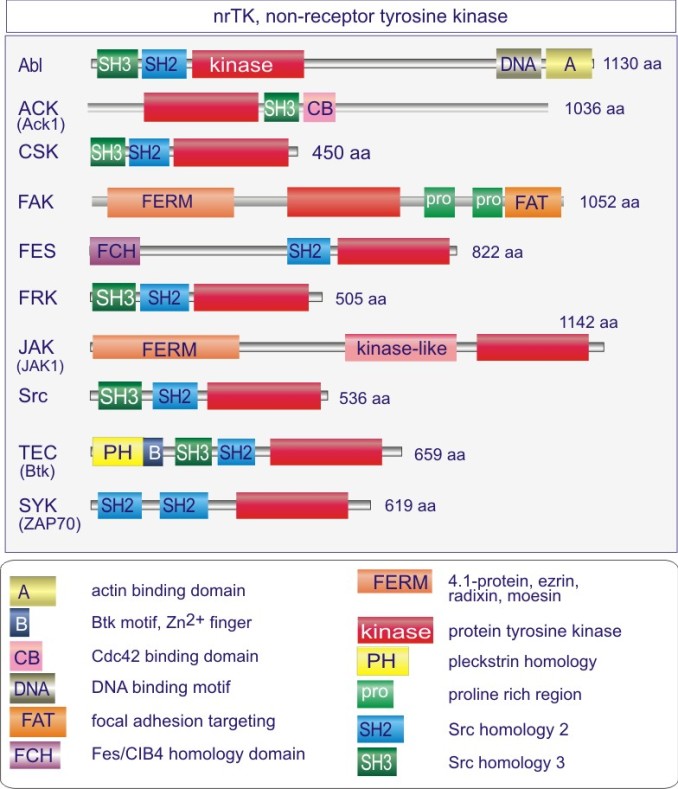
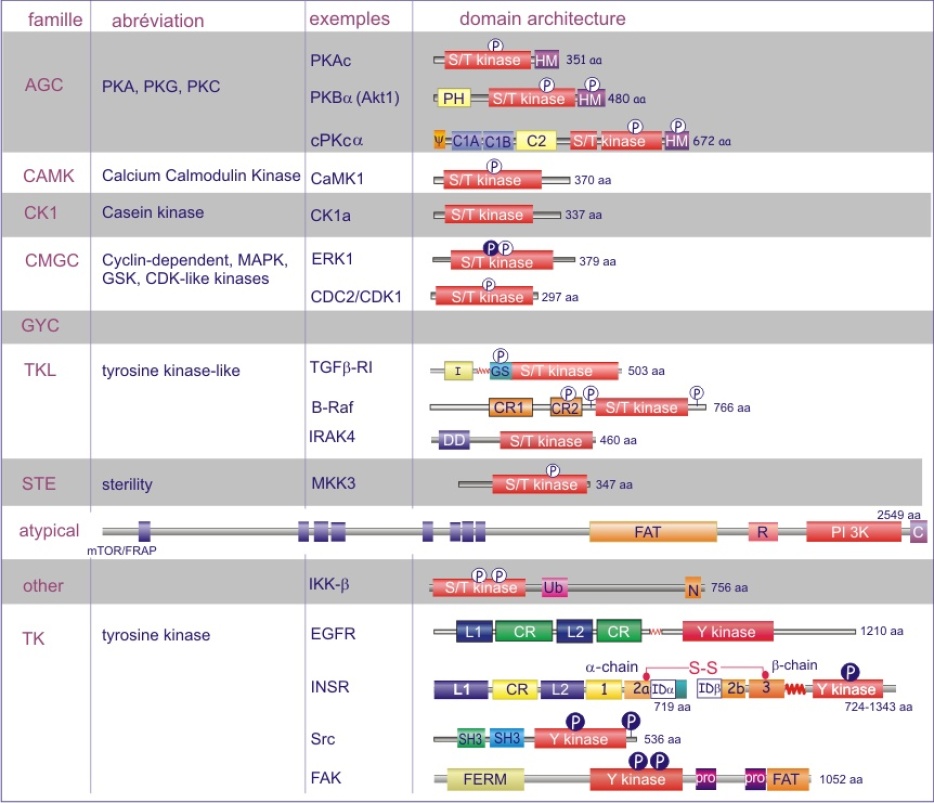
Les gènes codant les protéines kinases sont remarquablement abondants : 116 chez la levure Saccharomyces cerevisiae, 409 chez le nématode Caenorhabditis elegans (petit ver) et probablement environ 530 chez les vertébrés. Des analyses phylogénétiques de la séquence ont établi que le domaine catalytique de la plupart des sérine/thréonine kinases a un gène commun qui a donné naissance, par duplication et modification, à une douzaine de familles très tôt au cours de l'évolution (voir figure 18 pour les familles de protéine kinases et quelques-uns de leurs membres). Les domaines catalytiques conservés sont entouré par des séquences amino-acidiques plus variables qui déterminent à la fois l'affinité pour le substrat, l'interaction avec une sous-unité régulatrice ou, encore, la localisation cellulaire de la kinase.
Les histidine kinases, présentes chez la bactérie et la levure mais pas chez les nématodes, ont une origine distincte et leur domaine catalytique se replie différemment. Cette forme « primitive » ne sera pas évoquée plus avant dans cette ressource.
Les protéines kinases elles-mêmes sont sujettes à régulation ; elles doivent subir des modifications qui a) assurent le bon positionnement de l'ATP et b) les rendent accessibles à leur substrat et qui s'effectuent de différentes façons.
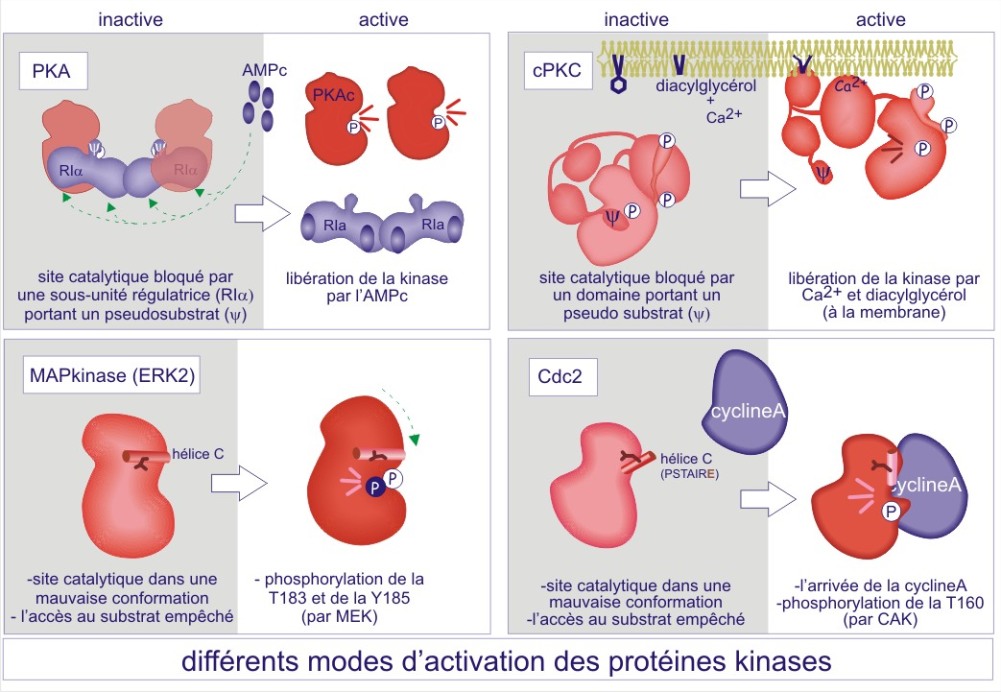
Par exemple, la protéine kinase A est composée d'une unité catalytique et d'une unité régulatrice bloquante. Cette dernière doit être éliminée par l'AMPc pour que l'enzyme puisse accéder au substrat (enzyme active). D'autres protéine kinases possèdent une séquence « pseudosubstrat», qui mime le « vrai substrat », mais qui par manque d'une sérine ou d'une thréonine ne peut être phosphorylée. L'interaction entre l'enzyme et son substrat est donc empêchée (le cas de CaMK, PKG et PKC). Il faudra respectivement l'association de Ca2+/calmoduline, de GMPc ou de diacylglycérol pour changer la conformation qui résultera en un déplacement du pseudosubstrat. Dans d'autres cas, parmi lesquels le récepteur de l'insuline et la MAPkinase, c'est la phosphorylation de la boucle d'activation dans la fente catalytique, qui rend possible l'accès au substrat. A l'inverse, pour les membres de la famille de la protéine kinase Src, c'est la déphosphorylation d'une tyrosine-phosphate, localisée loin du site catalytique, qui « ouvre » l'enzyme et la rend accessible au substrat. Pour les kinases régulant le cycle cellulaire, les cyclin dependent kinases (Cdk), ce sont des cyclines qui jouent le rôle de sous-unités activatrices en rendant la fente catalytique accessible au substrat substrat (voir figures 19A et 19B).
Certaines protéine-kinases, comme la protéine kinase A, ont un spectre d'activité très large alors que d'autres, telles que MEK (MAPK-ERK kinase) ou MLCK (myosin light chain kinase), sont très spécifiques d'un substrat donné. Dans le cas des kinases à large spectre, la question recurrente de savoir comment leur activation peut résulter en une réponse spécifique, reste en suspens. Cependant, la mise en évidence de protéines associées aux protéines kinases qui jouent un rôle dans leur localisation cellulaire précise (compartimentalisation), permet d'envisager l'existence d'un principe de régulation spatiale de l'activité, basé sur la présence ou l'absence, des facteurs d'activation et du substrat à phosphoryler. Ceci est également valable pour les protéine-phosphatases.
![]() Pour en savoir plus :
Pour en savoir plus :
Nous suggérons l'excellente revue de Louise Johnson et Richard Lewis sur la base structurale du contrôle de l'activité des protéines par la phosphorylation chez les eukaryotes et les prokaryotes.
« Phosphorylation control Johnson [pdf] » (1,9 Mo).
![]() Un article qui montre l'importance de la régulation spatiale des enzymes :
Un article qui montre l'importance de la régulation spatiale des enzymes :
« Compartimentalisation PKA b-AR Tsien [pdf] » (368 ko).
Complément : Excursion 5 : Mécanismes d'activation des protéines kinases.
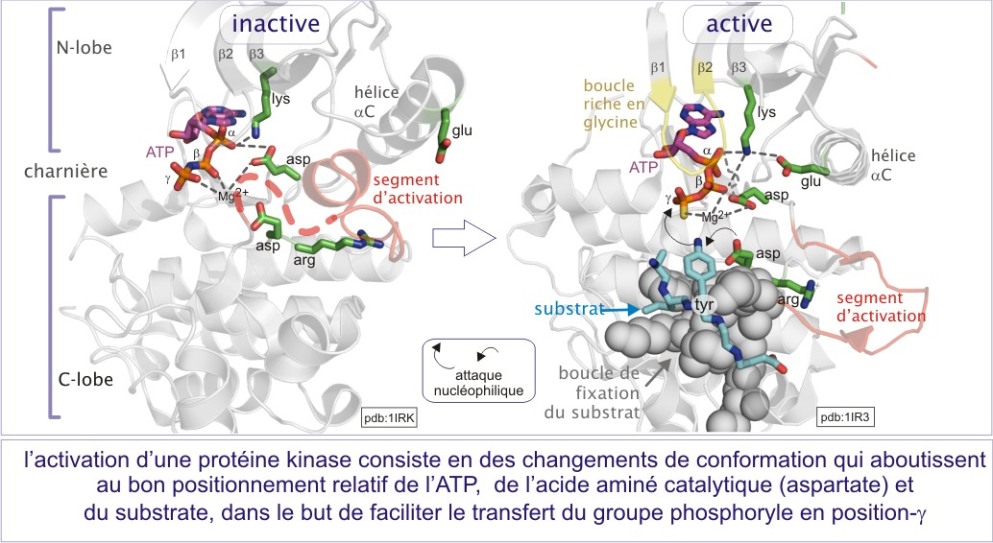
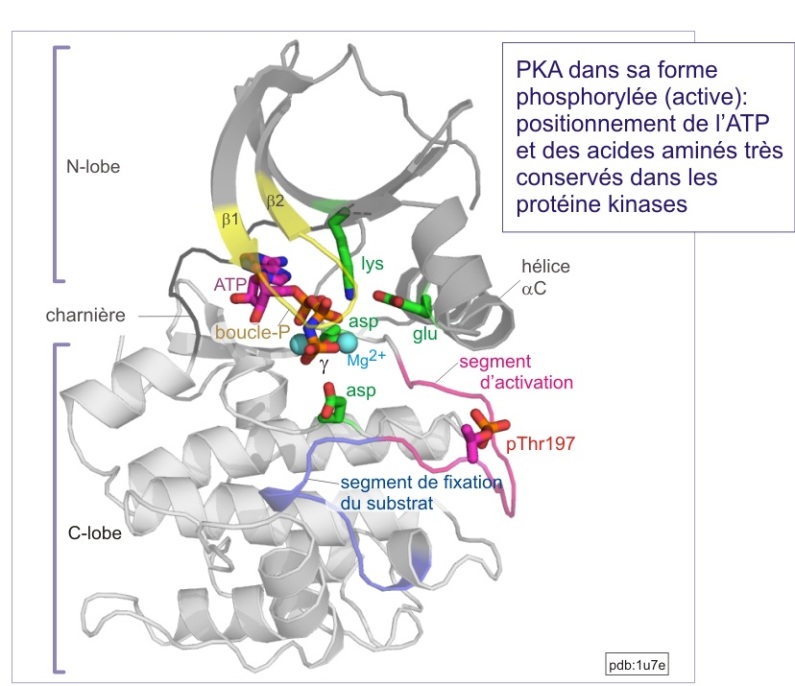
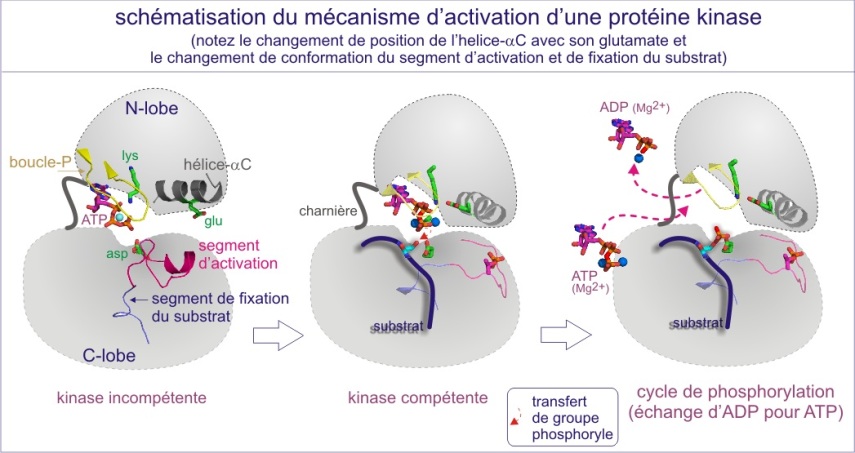
En première approximation, les sérine/thréonine ou tyrosine kinases ont toutes les mêmes repliements (« fold ») de leur domaine catalytique. Il consiste en 1) un lobe N-terminal, essentiellement formé d'un feuillet-\(\beta\) à cinq bandes et d'une hélice-\(\alpha\) , et 2) un lobe C-terminal, essentiellement composé d'hélices a et d'un élément essentiel, la région flexible (segment d'activation) impliquée dans la régulation de l'accès au substrat. La fente catalytique est à l'interface des deux lobes. La position de l'ATP est déterminée par une boucle fixant le phosphate (P-loop), très conservée au cours de l'évolution. Cette boucle s'aligne parallèlement à la fente catalytique et consiste en une séquence riche en glycine suivie d'une lysine et d'une sérine (ou d'une thréonine). On trouve cette boucle également dans les protéines liant le GTP.
L'activation nécessite à la fois l'accès du substrat à la fente catalytique et un positionnement de l'ATP favorable à son transfert (voir figure E10 et E11). De nombreuses protéines kinases doivent être phosphorylées sur un résidu thréonine ou tyrosine du “segment d'activation” (nommé aussi “boucle d'activation” ou “T-loop” dans le cas précis de Cdk2). Nous verrons le mécanisme général d'activation avec les exemples détaillés de trois types de protéines kinases :
Cdk2, sérine/thréonine kinase impliquée dans la régulation de la progression du cycle cellulaire.
ERK2, sérine/thréonine kinase activée par les mitogènes (facteurs de croissance)
Src, tyrosine kinase qui sous sa forme constitutivement active est impliquée dans la formation de tumeurs (sarcomes chez le rat).
Régulation de Cdk2
Les kinases dépendant des cyclines coordonnent la progression de la cellule eucaryote lors de son cycle cellulaire (prolifération cellulaire). Elles appartiennent à une sous-famille de sérine/thréonine kinases, Cdc2/CdkX, comprenant Cdk1 à 10 et des kinases apparentées à Cdk, telles que PCTK1 à 3.
Leur activation se fait en deux temps. D'abord il y liaison à une sous-unité régulatrice, la cycline, et ensuite, une phosphorylation par la kinase CAK (Cdk-activating kinase aussi appelée Cdk7) sur le résidu thréonine-160 du segment d'activation. Dans le cas de Cdk2 , la sous-unité régulatrice est la cyclineA ( CCNA1 ) et le complexe actif Cdk2/cylineA coordonne la progression à travers la phase de synthèse (phase S), pendant laquelle l'ADN est répliqué. L'analyse de structure par rayons X de la forme inactive de la kinase, montre que le segment d'activation gène partiellement le site de fixation d'ATP et que l'hélice a du lobe N-terminal, qui contient le motif PSTAIRE, est mal positionnée pour fixer les phosphates de l'ATP (voir figure E10). En absence de cycline A, la phosphorylation de la Thr-160 n'induit qu'un léger changement de structure et en conséquence, n'a que peu d'effet sur l'activité kinasique (0,3% de l'activité maximale). La seule association de la cyclineA, sans qu'il y ait phosphorylation, conduit à des changements significatifs de conformation : ouverture de la structure bilobaire, déplacement du segment d'activation avec exposition de la Thr-160 et glissement et rotation de 90° de l'hélice a du lobe N-terminal. Ces changements n'ont également que peu d'effet sur l'activité kinasique (0,3% de l'activité maximale). La liaison de la cyclineA et la phosphorylation de la Thr-160 procure 100% d'activité. Elles s'accompagnent du déplacement de la Thr-160 dans la molécule Cdk 2 en raison de trois nouveaux contacts avec : l'Arg50 (dans le motif PSTAIRE), l'Arg126 (voisin de l'aspartate catalytique) et l'Arg150. On peut considérer la Thr160 comme un centre organisateur qui assure un changement de conformation du segment d'activation dans la région comprise entre les résidus 152 et 163. Ce changement de conformation est crucial pour la reconnaissance du substrat peptidique HHAS*PRK.
NB : Il n'y pas de changement de conformation de la cyclineA lors de sa liaison à Cdk2.

L'inactivation de Cdk2 se fait par la perte de la cyclineA et la déphosphorylation de la Thr-160 par la phosphatase associée à la kinase (KAP, kinase associated phosphatase) . KAP ne peut pas déphosphoryler la pThr-160 du complexe, car le phosphoryle (P) n'est pas accessible. KAP n'est efficace que pour la pCdk2 seule.
Les kinases cyline-dépendantes sont soumises à un contrôle situé à un autre niveau : la phosphorylation et la déphosphorylation de la tyrosine-15, respectivement par la protéine kinase Wee1 puis par la phosphatase Cdc 25 ( MPIP1 ) (voir figure E11). En ce qui concerne Cdk2, les analyses structurales n'ont pas mis en évidence d'effet majeur sur la structure globale du complexe et il a donc été postulé que c'est l'encombrement stérique dû à la tyrosine phosphorylée qui prévient l'accès du substrat.
NB :
Wee signifie « petit » en Ecossais, c'est le phénotype de la levure qui se divise malgré sa petite taille due à son incapacité à arrêter le cycle cellulaire en phase G2/M (expression d'un mutant dit défectif).
Cdc signifie “cell division cycle”, protéines impliquées dans la régulation du cycle cellulaire.
Régulation de la MAPkinase
Les MAP kinases (mitogen activated protein kinases) relaient le signal membranaire vers le noyau. Elles furent découvertes pour leur activité kinasique en réponse à l'EGF ou l'insuline administrés à des fibroblastes swiss 3T3. Le terme de MAP kinase est maintenant réservé à une sous-famille de sérine/thréonine kinases comprenant 14 membres, MK1 à 14. Cette sous-famille est sous-divisée, en ERK1-5, p38 a-d kinase, JNK1-4 (c-Jun N-terminal kinase) et NLK (Nemo-like kinase), sur la base de la séquence des sites de phosphorylation du segment d'activation (soit respectivement LTEY, MTGY et MTPY) (voir figure E13). La MAPkinase découverte à l'origine fut renommée ERK pour « extracellular signal-regulated kinase » dont 5 variants existent. Les membres de la sous-famille des MAPkinases sont tous activés par une kinase de double spécificité, c'est-à-dire capable de phosphoryler aussi bien le résidu tyrosine que les résidus sérine et thréonine. Dans le cas d'ERK2, kinase que nous étudierons plus en détail ci-dessous, la kinase activante est MEK pour MAPkinase- ERK- Kinase.
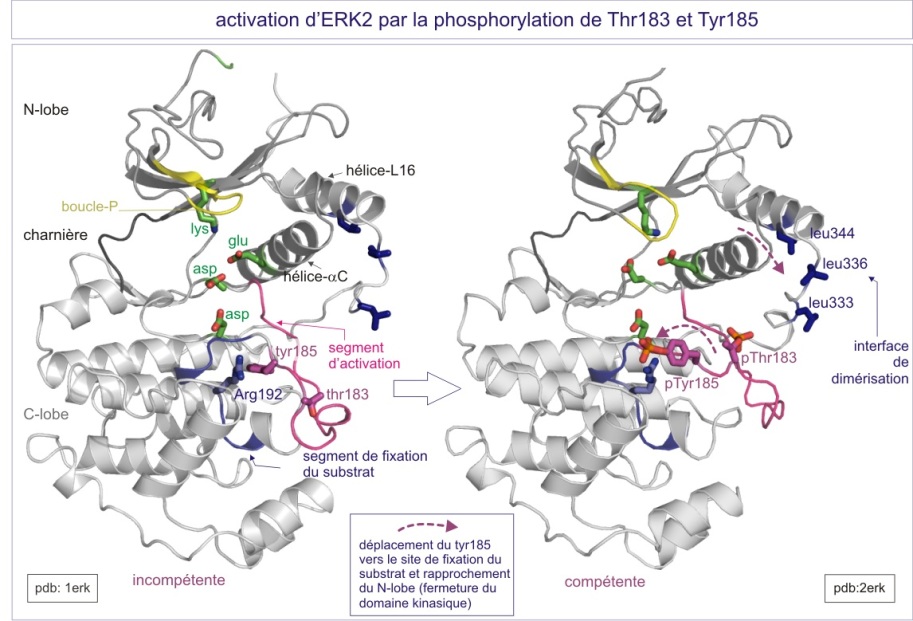
ERK2 atteint son activité maximum seulement quand la Thr183 et la Tyr185 du segment d'activation sont phosphorylées. La Thr183 est l'équivalent de la Thr160 dans le Ckd2 (et de la Thr197 dans la protéine kinase AMPc-dépendante (cAPK ou PKA)). Les formes monophosphorylées d'ERK2 ont moins de 1% de l'activité de la kinase biphosphorylée. L'ERK2 non-phosphorylée a un domaine kinasique à conformation ouverte, mais une fente catalytique bloquée par la Tyr185 dans un segment d'activation par ailleurs bien structuré. ERK a une extension C-terminale qui couvre à la fois les lobes C- et N-terminal et finit par une hélice a (nommée L16) qui entoure l'hélice-C (hélice du lobe N-terminal, proche de la fente catalytique) . Après phosphorylation, la pThr183 s'oriente vers l'intérieur de la protéine grâce à des contacts avec les groupes guanidium de l'Arg146 (voisin de l'aspartate catalytique) et de l'Arg170 (du segment d'activation). Ces contacts sont les équivalents de ceux de la Thr160 dans le cas de Cdk2/cyclineA. Après phosphorylation, la pTyr185 se déplace vers la surface de la protéine où elle interagit avec l'Arg189 et l'Arg192. La phosphorylation impose une réorientation du segment d'activation située entre la Gly167 (DFG motif) et l'Ala187. Le changement de position de l'Ala187, qui est maintenant liée à l'Arg192, est crucial pour la création d'un motif proline capable de reconnaître le substrat (sur ses résidus adjacents à la sérine et la thréonine).
Dans le cas d'ERK, la double phosphorylation aboutit au même effet (rendre la kinase catalytiquement compétente) que celui résultant de l'association de la cyclineA puis de la phosphorylation de la Thr160, lors de l'activation de la Cdk2 ; 1) la reconformation plus fermée du domaine catalytique, permettant 2) le bon positionnement des résidus nécessaires à la liaison à l'ATP et à la réaction chimique (résidu catalytique) et 3) une réorientation du segment d'activation qui crée un site correct de reconnaissance du substrat.
L'ERK2 activé se dimérise grâce au repositionnement de l'hélice L16, qui expose maintenant trois résidus leucine (333, 336 et 344) capables de se lier aux leucines d'une seconde ERK activée par la formation d'une “leucine zipper” (fermeture éclair faite de leucines) . En conséquence la constante de dissociation (Kd) passe 20 m M pour ERK2 non-active à 7,5 nM pour 2pERK2-active. Enfin, la 2pERK-dimérisée est reconnue par la machinerie d'import nucléaire (par un signal de localisation nucléaire (NLS) non encore identifié).
Inhiber la protéine kinase ; faut-il cibler son état actif ou inactif ?
L'analyse structurale de plusieurs sérine/thréonine kinases révèle qu'à l'état inactif, elles présentent une grande variabilité. A l'état actif au contraire, ces différentes structures convergent vers un cadre commun d'organisation, en ce qui concerne l'orientation correcte des acides aminés du site catalytique et du segment d'activation. Ceci a des conséquences importantes pour la mise au point d'inhibiteurs kinasiques à but thérapeutique (anti-cancéreux par exemple). Pour ce qui est des protéines kinases inactives, des inhibiteurs hautement sélectifs ont été obtenus (qui ciblent le site catalytique). En revanche ce n'est pas le cas pour les kinases dans leur conformation active car leurs sites catalytiques se ressemblent énormément. Cependant, un important inconvénient du ciblage des kinases inactives vient du fait qu'elles échappent « facilement » à l'inhibiteur car sujettes à de fréquentes mutations. Du point de vue de leur fonctionnement, les mutations ponctuelles sont aisément tolérées comme ceci est reflété par la grande variabilité de la structure du domaine catalytique des kinases inactives. Ces mutations qui n'ont pas de conséquences fonctionnelles majeures, sont le plus souvent à l'origine d'une perte considérable d'affinité pour l'inhibiteur (cas de l'oncogène Bcr-Abl ( Abl-1 ) et l'interaction avec son inhibiteur Gleevec®
Cliquer ici pour plus d'information sur Gleevec et leucémie myloïde chronique.
Régulation de Src
Src , pour s a rc ome (tumeur de tissu mou), est le prototype d'une famille de tyrosine protéine kinases qui comprend les membres, Blk, Lck , Fgr, Fyn , Hck, Lyn, Src , Srms , Frk et Yes . Src a été découvert comme un produit viral (Src viral ou v-Src) qui entraine la transformation de tissu mou chez le poulet. Plus tard, chez l'homme, un équivalent fut mis en évidence et dénommé c-Src. L'analyse de son mode d'action est à l'origine de la découverte de la phoshorylation de la tyrosine (et son rôle dans la transformation cellulaire).
La protéine kinase Src comprend cinq domaines:
un domaine N-terminal unique impliqué dans la localisation sub-cellulaire (par exemple attachement membranaire par ;
un domaine SH3 ;
un domaine SH2 ;
un domaine kinasique
un domaine C-terminal portant une tyrosine en position 527. La Tyr527 est un substrat d'une autre tyrosine protéine kinase, la Csk (c-Src kinase) dont la phosphorylation inhibe Src. La forme oncogénique, v-Src, est dépourvue de ce site de phosphorylation ce qui entraîne son activité dérégulée, base de la transformation cellulaire (voir figure E13).
La tyrosine phosphorylée (pTyr527) de l'extrémité C-terminale est à 4 nm du site catalytique, et n'agit donc pas directement sur le segment d'activation. Par ailleurs, la pTyr527 se comporte comme un site de fixation du domaine SH2. La liaison intramoléculaire ainsi réalisée rend le domaine catalytique inactif de deux façons : d'une part, elle apporte une contrainte à l'hélice-C ( proche de la fente catalytique) , qui ne peut plus se positionner dans une conformation active et d'autre part, elle lie les domaines SH3 et SH2 aux extrémités des deux lobes du domaine catalytique et, ce faisant, elle contrarie sa conformation active (« clamp SH2/SH3 » comparable au «sabot de Denver »). La tyrosine kinase ainsi empêchée ressemble par sa structure à la Cdk2 non liée à la cyclineA (les deux kinases sont inactives)
NB :
La liaison du domaine SH2 avec la pTyr527 se fait par des ponts hydrogène établis avec l'Arg155, leArg175, le Glu78 (par le groupe NH de l'axe polypeptidique) et avec la Ser177 et la Thr79 (par l'atome O g ).
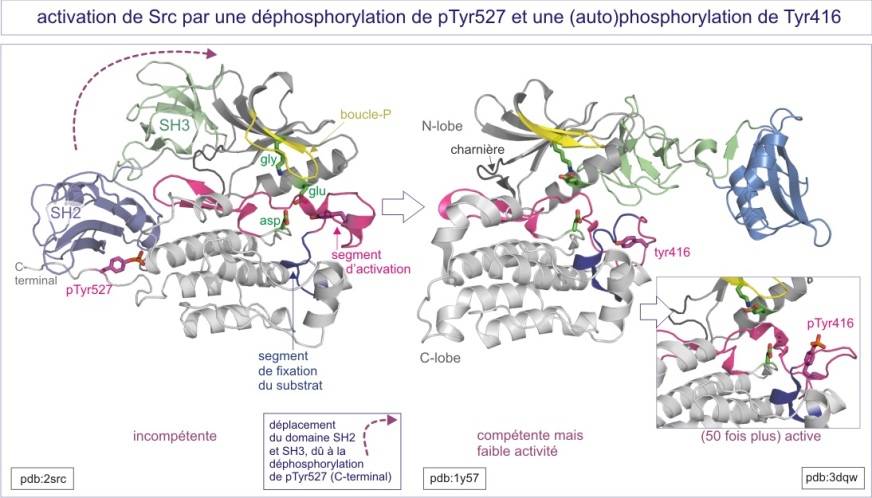
Comme nous le verrons dans la section traitant du récepteur à l'EGF, le domaine SH2 interagit préférentiellement avec deux acides aminés, une phosphotyrosine et un amino acide dit de «spécificité » localisé à pTyr+3 (asparagine, isoleucine ou méthionine par exemple). Cet amino acide de “spécifité » fait défaut dans le domaine C-terminal de Src ce qui conduit à une liaison relativement faible. Une conséquence importante de cette faible liaison est que n'importe quelle protéine portant une tyrosine phosphate plus un amino acide de « spécificité » favorable, peut remplacer le domaine SH2 intramoléculaire, « ouvrir » la protéine et donc enlever le clamp SH2/SH3, ce qui conduit à l'activation de la Src (voir figure E14).
L'interaction entre le domaine SH3 et le lobe N-terminal du domaine catalytique est rendue possible par la présence d'une conformation « polyproline » (bien qu'une seule proline soit présente dans ce cas particulier) qui sert de site d'interaction avec le domaine SH3. La leucine de ce site mérite une mention spéciale puisqu'elle interagit avec le lobe N-terminal du domaine catalytique et est responsable de sa conformation clampée. Le remplacement de la Leu255 par la Val (plus petite), augmente l'activité kinasique qui n'est plus sensible à la phosphorylation de la Tyr527.
Le segment d'activation de Src, compris entre les motifs DFG et APE, se situe entre les résidus 404 et 432. Les résidus 404-411 s'agencent en une hélice de type 3 10 , similaire à l'hélice L12 dans CDK2 (résidus 145-152). Les résidus hydrophobes de l'hélice du segment d'activation interagissent avec l'hélice-C (du lobe N-terminal) et la stabilise dans sa conformation inactive, c'est-à-dire incapable de bien positionner les groupes phosphoryles de l'ATP. Plus avant dans le segment d'activation, les résidus 413-418 forment une hélice qui positionne la tyr416 près de la boucle catalytique et, en prenant la place du substrat, bloque le site catalytique. Lorsque le clamp SH3/SH2 est enlevé (par une tyrosine phosphatase ou par compétition avec un autre domaine SH2 (externe)), l'hélice-C se positionne correctement et permet un bon placement du groupe b -phosphoryle de l'ATP. La trans-autophosphorylation de la Tyr416 s'ensuit, repositionnant ainsi le segment d'activation. L'activité kinasique est multipliée par 50.
![]() Pour en savoir plus sur Src et la découverte de la phosphorylation de tyrosine :
Pour en savoir plus sur Src et la découverte de la phosphorylation de tyrosine :
« Src protein purif Brugge [pdf] » (805 Ko).
« Src tyrosine Sefton [pdf] » (1,64 Mo).
Complément : Excursion 6 : Krebs et Fischer découvrent la phosphorylation de la phosphorylase a.
Dès 1940, grâce au travail d'Arda Green et du couple Carl et Gerty Cori, il était bien établi que la dégradation du glycogène dans le muscle squelettique, et d'autres types de cellules, s'effectuait par un processus de phosphorylyse (glycogène (n) + ATP ---> glycogène (n-1) + glucose-1-P + ADP), catalysé par l'enzyme nommée phosphorylase. Purifiée à partir du muscle de lapin, l'enzyme fut caractérisée sous deux états fonctionnels, la phosphorylase a et phosphorylase b qui différaient sur deux points : 1) seule la phosphorylase a pouvait être purifiée sous forme cristalline et 2) contrairement à la phosphorylase a, l'activité de la phosphorylase b était faible et nécessitait la présence de forte concentrations de 5'-AMP pour se manifester.
Par la suite, les Cori découvrirent une enzyme capable, in vitro, de convertir la phosphorylase a (active) en phosphorylase b (moins active). Parce qu'ils pensaient que le 5'-AMP était le groupe prostéthique responsable de l'activité de l'enzyme, ils nommèrent l'enzyme de conversion « PR enzyme », pour Prosthetic group Removing enzyme. L'attribution erronée de ce rôle au groupe prosthétique était justifiée vu que la notion d'allostérie et donc le changement de l'activité dû à la fixation des facteurs aux sites non-catalytiques de l'enzyme, n'avait pas encore été formulée. On sait maintenant que l'enzyme de conversion est la sérine/thréonine phosphatase PP1G.
En automne 1953, Fisher et Krebs commençèrent à travailler ensemble dans le nouveau Département de Biochimie, University of Washington, à Seattle, sur le mécanisme encore inconnu de l'effet stimulant de 5'-AMP sur la phosphorylase b et de son inefficacité sur la phosphorylase a (forme active). Au cours de leurs premiers essais, ils échouèrent dans leur tentative de purifier, par cristallisation selon la procédure de Cori, la phosphorylase a. Plus étonnant était l'observation que dans son état partiellement pur, la phosphorylase était entièrement dans la forme b (peu active). Selon le propos de Krebs :
“Autant que nous ayons pu le déterminer, nous avions très fidèlement suivi la procédure de Cori sauf que nous avions décanté l'extrait cru de muscle par centrifugation plutôt que par filtration sur papier. Bien que ceci semblait être un changement mineur, nous avons néanmoins inclus l'étape de filtration dans notre préparation ultérieure de la phosphorylase. A notre grande surprise, cette fois nous obtînmes de la phosphorylase a facilement cristallisée, telle qu'elle devait être. Deux conclusions pouvaient être tirées pour expliquer les résultats. Primo, les extraits de muscle « en repos » doivent contenir la phosphorylase surtout dans sa forme b (peu active) plutôt que dans sa forme a, comme l'avait postulé le couple Cori. Secundo, la filtration sur papier de l'extrait de muscle « en repos » doit déclencher la conversion in vitro de la phosphorylase b en phosphorylase a ».
(discours Nobel de E. Krebs).

Fischer et Krebs montrèrent par la suite que lorsqu'on lavait soigneusement le papier filtre ou qu'on laissait l'extrait cru de muscle « vieillir » quelques heures sur la paillasse, la phosphorylase a (active) était totalement éliminée. Il s'avèra que le composant critique présent dans le filtre était le Ca2+ et que le composant perdu lors du vieillisement était l'ATP. Sachant donc que l'ATP est nécessaire à la conversion Ca2+-dépendante de la phopshorylase b (peu active) en phosphorylase a (active), ils postulèrent qu'une réaction de phosphotransférase était impliquée. En effet, une « enzyme de conversion » est requise durant le processus d'activation de la phosphorylase, ADP était le second produit de réaction. Enfin, ils démontrèrent que le passage de phosphorylase b à phophorylase a (active), nécessite la phosphorylation de la sérine-14, rendue possible par « la phosphorylase kinase ».
Revenant sur l'existence de l'enzyme PR, antérieurement découverte par les Cori, ils démontrèrent que cette enzyme est capable de libérer le phosphate inorganique (Pi ou H 2 PO4 - ) et peut donc s'identifier en tant que phosphorylase phosphatase (PP1G). Dès ce moment, on a pu considérer que phosphorylation et déphosphorylation sont des processus capables d'altérer l'activité enzymatique. En d'autres termes, la phosphorylation peut être rangée parmi les modifications allostériques.
Dans la fin de la décennie 1950 on pouvait écrire les équations d'interconversion des phosphorylases musculaires, comme suit :
phosphorylase kinase
Phosphorylase b (dimère peu actif) + 2ATP ---> phosphorylase a (dimère actif) + 2ADP
phosphorylase phosphatase
Phosphorylase a (dimère actif) + 2H20 ---> phosphorylase b (dimère peu actif) + 2Pi
Articles historiques :
Krebs, EG and Fischer EH. The phosphorylase b to a converting enzyme of rabbit skeletal muscle. Biochim. Biophys. Acta 1956 ;20 :150-157.
Edmond H. Fischer and Edwin G. Krebs. Biochimica et Biophysica Acta, Volume 1000, 1989, Pages 297-309
Les travaux de Carl et Gerty Cori furent récompensés par l'attribution du prix Nobel de Physiologie ou Médecine en 1947 “for their discovery of the course of the catalytic conversion of glycogen” (partagé avec Bernardo A Houssay “for his discovery of the part played by the hormone of the anterior pituitary lobe in the metabolism of sugar").
Les travaux d'Edmond Fischer et Edwin Krebs furent récompensés par l'attribution du prix Nobel de Physiologie ou Médecine en 1992 “ for their discoveries concerning reversible protein phosphorylation as a biological regulatory mechanism ».
Complément : Excursion 7 : Familles des kinases
Cette excursion montre des tableaux récapitulant en détail les différentes familles de protéine kinases ainsi que l'architecture des domaines protéiques.