Chapitre 2 : Les réactions chimiques à l'interface air-eau
Rappel :
Rappels de thermodynamique
Définition : Notion de spontanéité
Un processus spontané est un processus qui se produit sans action extérieure dans un système laissé à l'abandon. Un exemple simple de processus spontané est la formation de rouille sur une pièce métallique. La formation de rouille se produit spontanément de façon continue, elle s'écrit par la réaction : 4 Fe(s) + 3 O2(g) = 2 Fe2O3(s). La quantité de Fe(s) décroît et la quantité de rouille (Fe2O3(s)) croît jusqu'à ce qu'un état d'équilibre final soit atteint, dans lequel toute le Fe(s) a été transformé en Fe2O3(s). Cette réaction est dite spontanée.
Concernant la notion de spontanéité, on peut énoncer que :
-lorsqu'un processus est spontané, le processus antagoniste est non spontané
-à la fois les processus spontanés et non spontanés sont possibles, mais seulement le processus spontané se produira sans intervention.
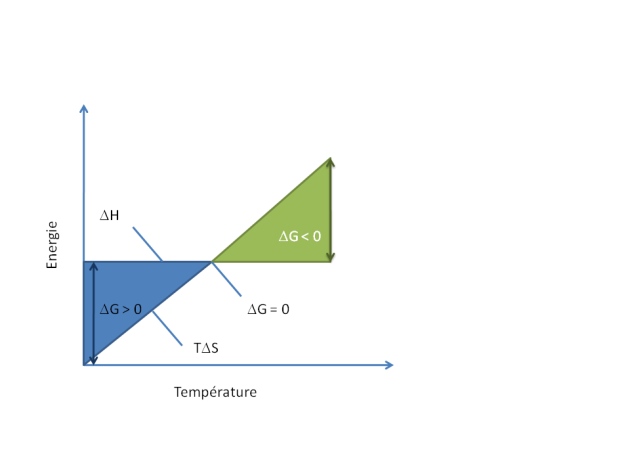
D'un point de vue thermodynamique, la seule considération de la variation d'enthalpie ne suffit pas déduire si le système est spontané ou pas. Le concept d'entropie est un concept thermodynamique qui permet de rendre compte des propriétés thermodynamiques relatives à la façon dont l'énergie d'un système est distribuée parmi les niveaux d'énergie microscopiques disponibles. La variation d'entropie est la différence d'entropie entre deux états d'un système. Dans le cas des processus spontanés, la variation d'entropie entre le système à l'état initial et à l'état final est systématiquement positive.
Définition : Critère de spontanéité
Le critère de spontanéité d'une réaction constitue l'énoncé du second principe de la thermodynamique selon lequel tout processus spontané provoque une augmentation de l'entropie de l'univers.
![]() Sunivers =
Sunivers = ![]() Ssystème +
Ssystème + ![]() Senvironnement > 0
Senvironnement > 0
D'après cette expression, lorsqu'un processus produit une variation d'entropie positive à la fois dans le système et dans son environnement, le processus est spontané. A l'inverse, si ces variations d'entropie sont négatives, le processus est non spontané.
Définition : Energie libre et variation d'énergie libre
La considération de l'entropie comme critère de spontanéité est difficile car pour estimer ![]() Sunivers il faut systématiquement évaluer
Sunivers il faut systématiquement évaluer ![]() Senvironnement , ce qui n'est pas toujours possible.
Senvironnement , ce qui n'est pas toujours possible.
Pour considérer les changements du système lui-même, il faut considérer la fonction thermodynamique énergie libre (G). G = H – TS. La variation d'énergie libre d'un système à T constant est ![]() G =
G = ![]() H - T
H - T![]() S. Dans cette équation tous les termes sont relatifs au système en question.
S. Dans cette équation tous les termes sont relatifs au système en question.
A pression et température constantes, lorsque
-![]() G<0 le processus est spontané
G<0 le processus est spontané
-![]() G=0 le processus est à l'équilibre
G=0 le processus est à l'équilibre
-![]() G>0 le processus est non spontané
G>0 le processus est non spontané
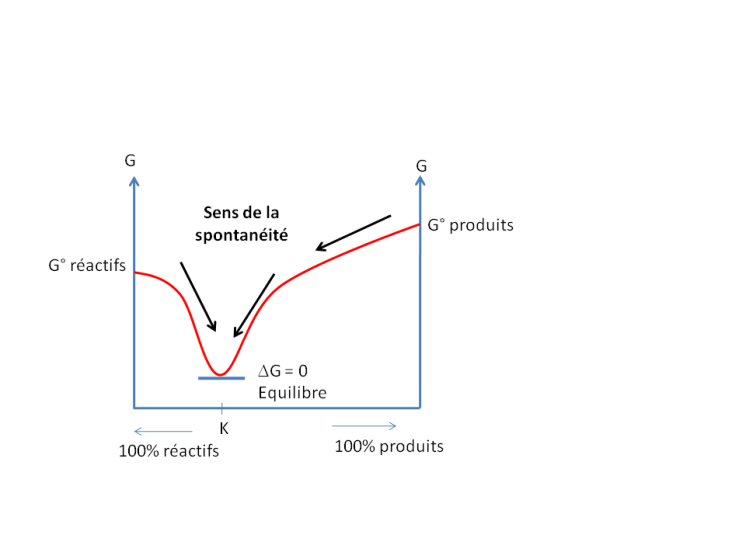
Lorsque l'équilibre d'une réaction est atteint, on peut écrire
![]() G =
G = ![]() G°+RT lnKeq = 0
G°+RT lnKeq = 0
avec K, la constante d'équilibre de la réaction
avec a : les activités des réactifs et produits de la réaction, et ![]() les coefficients stœchiométriques des réactifs et produits de la réaction.
les coefficients stœchiométriques des réactifs et produits de la réaction.
Les interactions chimiques entre les différents compartiments d'un cycle concernent uniquement des réactions spontanées.
Définition : Fraction molaire et pression partielle
L'air sec est constitué de trois gaz principaux, dont les proportions restent constantes : N2 (78% en volume), O2 (21% en volume), Ar (1% en volume). On trouve également du CO2 (environ 0,037%), des gaz rares (Ne, He, Kr, Xe) et des gaz réactifs tels que CH4, H2, les oxydes d'azote (N2O, NO2), CO, O3, SH2, SO2, NH3. Les gaz réactifs participent aux réactions dans les cycles biogéochimiques naturels. Leurs concentrations spatiales et temporelles varient peu. Les transferts entre les différents gaz constituants l'atmosphère sont régis par plusieurs phénomènes physico-chimiques.
La fraction molaire d'un gaz dans un mélange gazeux s'exprime par la relation :
La pression partielle d'un gaz dans un mélange est la pression exercée par ce gaz s'il occupait seul tout le volume. La pression totale correspond à la somme des pressions partielles des différents gaz du mélange. La pression partielle s'exprime par la relation (loi de Dalton) :
Définition : Tension de vapeur saturante de l'eau
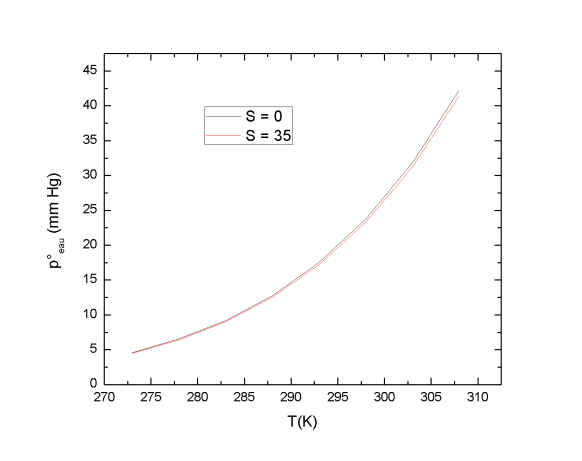
Le contenu en vapeur d'eau d'une atmosphère humide peut être caractérisé par sa pression partielle peau. Un équilibre s'établit entre une masse d'eau et un volume d'air sec. Des molécules d'eau se volatilisent pour envahir l'air jusqu'à ce qu'un équilibre entre l'eau et sa vapeur s'établisse. Ce processus de changement d'état se produit à température constante. La loi de Raoult permet d'obtenir la tension de vapeur d'un composé en fonction de sa fraction molaire et de sa tension de vapeur à une température donnée :
L'influence de la salinité du milieu sur la tension de vapeur est exprimée par l'équation suivante :
Avec S la salinité du milieu (sans unité) et T la température (K). La salinité du milieu influence peu la tension de vapeur saturante de l'eau. Par contre, une augmentation de température du milieu entraîne une augmentation de la tension de vapeur saturante de l'eau.
Définition : Solubilité des gaz
La dissolution d'un gaz dans l'eau suit la loi de Henry :
KHX représente le coefficient de solubilité (mol L-1 atm-1), également appelé constante de Henry.
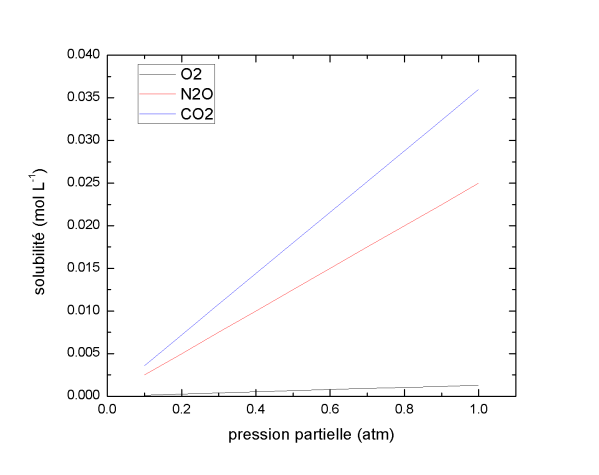
Les gaz tels que le CO2 et le N2O ont des coefficients de solubilité élevés, très supérieurs à celui d'O2. Dans des conditions de température et de pression partielle identiques, leur solubilisation dans l'eau sera donc supérieure à celle d'O2.
Influence de la température sur la solubilisation des gaz
La relation de van't Hoff permet de calculer à toute valeur de T la solubilité d'un gaz.
Pour mieux comprendre l'évolution de la solubilité d'un gaz en fonction de la température, intéressons nous à la dissolution du CO2(g) dans l'eau. La réaction de solubilisation de CO2(g) est la suivante :
CO2(g) + H2O(l) = H2CO3(l)
A partir des variations d'enthalpie standard de formation (à 25°C) des différents composés de la réaction (tableau 1), on peut calculer la variation d'enthalpie de la réaction de dissolution du CO2.
H2CO3 (l) | H2O (l) | CO2 (g) | |
| -692,0 | -285,8 | -393,5 |
KH (mol L-1 atm-1) | 3,6.10-2 |
![]() H =
H = ![]() H°(H2CO3) – (
H°(H2CO3) – (![]() H°(H2O) +
H°(H2O) + ![]() H°(CO2)) = -692,0-(-285,8-393,5) = -12,7 kJ.mol-1
H°(CO2)) = -692,0-(-285,8-393,5) = -12,7 kJ.mol-1
L'application de la relation de van't Hoff permet de calculer l'évolution de la constante de Henry en fonction de la température
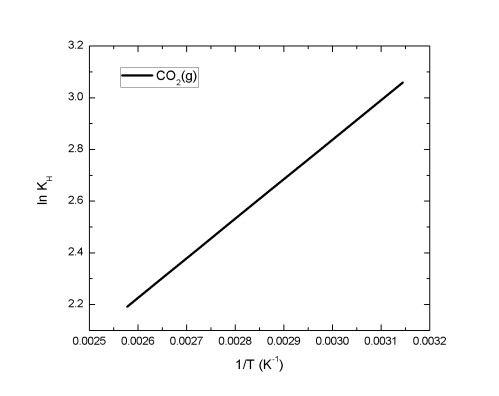
Dans l'intervalle de température de 20°C à 90°C, la solubilité de CO2 diminue lorsque la température augmente.
Le cas du CO2 peut être généralisé à tous les autres gaz. La solubilité d'un gaz diminue lorsque la température augmente.
Influence de la salinité sur la solubilisation des gaz
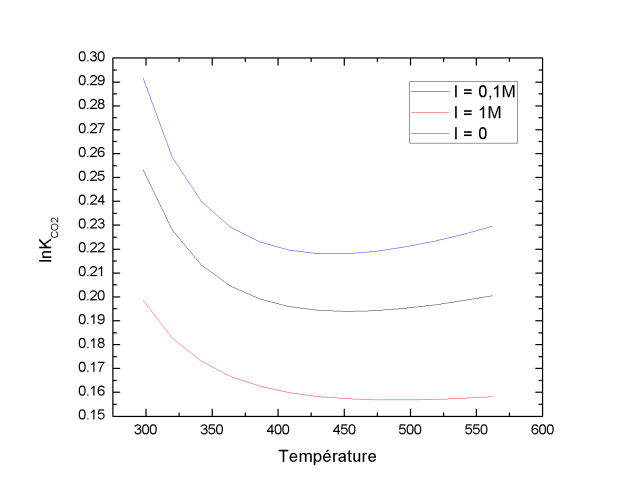
Le coefficient apparent de solubilité de CO2 en fonction de la température et de la salinité s'exprime par l'algorithme suivant :
![]()
Avec les coefficients a0 à a9 spécifiques au CO2 pour une pression égale à la pression de vapeur saturante du CO2. L'évolution du logarithme népérien de la constante apparente d'équilibre de solubilité de CO2 à pression constante en fonction de la température et de la force ionique est présentée dans le graphique 4.Ce graphique illustre bien que la solubilité du CO2(g) tend à diminuer lorsque la salinité du milieu augmente.
Définition : Réactions de transfert
L'étude d'un cycle biogéochimique ne peut se faire sans considérer la notion de transfert d'une substance d'un compartiment à un autre. La comparaison des constantes de cinétique relatives à un échange entre deux compartiments permet d'identifier si une espèce X aura un impact dans une zone plus ou moins éloignée de sa source. On considère qu'une espèce X aura un impact dans une zone donnée si le temps de transport séparant cette zone de la source est inférieur au temps de vie (![]() ) de l'espèce. Le temps de vie de l'espèce correspond au temps au bout duquel la concentration de l'espèce est divisée par 2,718 (e).
) de l'espèce. Le temps de vie de l'espèce correspond au temps au bout duquel la concentration de l'espèce est divisée par 2,718 (e).
Le temps de vie d'une espèce est défini par rapport à un processus ou une somme de processus d'élimination donnés, il dépend de la saison, de l'humidité, de la situation géographique car la constante de vitesse varie en fonction des paramètres physico-chimiques du milieu. Considérons le transfert d'un gaz du compartiment air vers le compartiment eau, ceci suppose que le taux d'évaporation est plus faible que le taux de condensation, soit K.CG < K'.pG (avec CG la concentration du gaz dans l'eau et pG la pression partielle du gaz). Le flux de gaz transféré est un nombre de moles par unité de surface et par unité de temps et est exprimé par l'équation 7.
Dans cette équation, K est appelé coefficient de transfert, il a la dimension d'une vitesse.
La quantité de gaz dans l'atmosphère étant infinie on peut considérer que sa pression partielle à la surface d'une eau est constante et qu'elle peut être considérée égale à la concentration à saturation C°G.
Si l'on exprime l'équation 8 de façon à faire apparaître la variation de concentration en gaz, on obtient l'équation suivante :
Dans cette équation, k a une dimension (temps-1).
Intéressons nous à l'évolution cinétique de la dissolution de l'oxygène dans une eau en fonction de la salinité. Pour une eau de mer (salinité 35) à 15°C la limite de solubilité de l'oxygène à P = 1 bar est de 8,13 mg.L-1.
L'intégration de l'équation 9 donne :
Avec a = CiG – C°G et b = C°G
Pour une constante de transfert de 0,8 j-1 et une concentration initiale de 0,5 mg.L-1, on obtient la courbe de cinétique de transfert suivante :
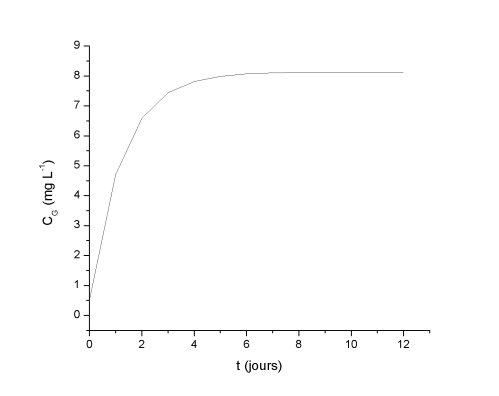
L'eau atteint son niveau de saturation en oxygène au bout de 5 jours.
Ainsi, nous avons mis en évidence que la solubilité d'un gaz à pression constante dépend de la température et de la salinité du milieu.
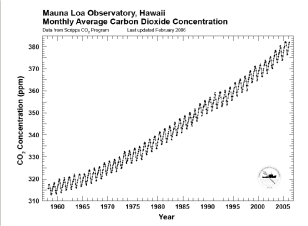
Le CO2 est indispensable à la vie car il constitue la seule source de carbone disponible pour les plantes (terrestres et aquatiques) dans la photosynthèse. L'effet de serre est un phénomène naturel indispensable à la vie sur Terre. L'atmosphère terrestre est transparent aux rayonnements UV et visibles du soleil. Ces rayonnements sont absorbés par la surface de la Terre et la réchauffent. Une partie de cette énergie absorbée est réémise sous la forme d'un rayonnement infrarouge. Certains gaz atmosphériques tels que le CO2 et la vapeur d'eau absorbent une partie du rayonnement infrarouge. L'énergie ainsi accumulée dans l'atmosphère provoque un effet de réchauffement : c'est ce que l'on appelle « l'effet de serre ». Cet effet de serre naturel est essentiel à la vie sur Terre puisqu'il y maintient des températures favorables. Sans cet effet de serre naturel, la Terre serait recouverte de glace. L'augmentation des activités industrielles (utilisation des combustibles fossiles en particulier, et déforestation qui tend à diminuer les « puits » de CO2) depuis le début du 19ème siècle a provoqué une augmentation des teneurs en CO2 atmosphérique, entraînant ainsi l'apparition d'un effet de serre dit « additionnel ». Cet effet de serre additionnel a pour conséquence d'augmenter la température moyenne à la surface de la Terre puisque les cinétiques des réactions de consommation du CO2 à l'intérieur d'un cycle ne sont pas suffisamment rapides pour absorber le surplus de CO2 produit. D'autre part, comme nous l'avons démontré précédemment, une augmentation de la température de l'eau entraîne la diminution de la solubilisation du CO2 dans les eaux de surfaces ainsi qu'une élévation de la tension de vapeur saturante de l'eau. Ce phénomène a pour conséquence la formation de nuages chargés d'eau dans l'atmosphère, capable d'absorber une partie de rayonnement solaires, limitant ainsi l'effet de serre. Lorsque la salinité du milieu est élevée, la solubilisation du CO2 est défavorisée. La ressource en eau de la Terre étant constituée à plus de 97% d'eau de mer, les transferts entre les gaz atmosphériques et les eaux salines sont peu favorisés lorsque la température des océans augmente, ce qui a pour conséquence un accroissement continu des teneurs en CO2 dans l'atmosphère (schema 7). Nous avons pris ici pour exemple le CO2 afin d'expliquer les transferts entre l'atmosphère et l'eau et expliquer les conséquences de l'effet de serre. Cependant, le CO2 n'est pas le seul gaz responsable de l'effet de serre. En effet, le méthane (CH4), l'ozone (O3) les oxydes nitreux et les chlorofluorocarbones (CFC) sont des gaz dont les capacités d'absorption du rayonnement infrarouge sont bien plus élevées que celle du CO2. De plus les concentrations dans l'atmosphère de ces gaz croissent beaucoup plus rapidement que les concentrations en CO2.
Définition : Réactions acide-base
D'après la définition de Brönsted, un acide est un donneur de proton et une base est un accepteur de proton. Dans la réaction NH3 + H2O = NH4+ + OH-, H2O est un acide qui cède un proton à NH3, qui se comporte comme une base en acceptant ce proton. NH3 est un acide faible, la réaction opposée à celle que nous venons d'étudier est possible. Dans le sens gauche-droite de la réaction, NH4+ est un donneur de proton (donc un acide) et OH- est un accepteur de proton (donc une base). Les couples NH3/NH4+ et H2O/OH- sont des paires conjuguées, NH4+ est l'acide conjugué de NH3 et OH- est la base conjuguée de H2O.
- Notion d'acide (base) fort(e) et d'acide (base) faible
Soit l'équilibre en solution aqueuse AH + H2O = A- + H3O+ avec AH l'acide et A- sa base conjuguée.
La constante d'équilibre associée à cette réaction prise dans le sens gauche-droite est :
Cette constante est appelée constante d'acidité. On peut également définir de la même façon la constante de basicité de la réaction si l'on considère le sens gauche-droite de la réaction, mais de manière générale ce sont les valeurs de Ka qui sont utilisées.
La valeur de la constante d'acidité d'une réaction permet de donner une indication sur la force d'un acide ou d'une base. Ainsi, plus la valeur de Ka est élevée, plus l'acide du couple sera fort (et donc plus la base conjuguée sera faible). A l'inverse, plus la valeur de Ka est faible, plus l'acide du couple est faible (et donc plus la base conjuguée est forte). Compte tenu de la force des acides et des bases, et de la valeur de Ka du couple considéré, dans le cas d'un acide fort (Ka élevé) l'équilibre réactionnel sera fortement déplacé dans le sens de dissociation de l'acide. Pour cette raison, la réaction est considérée totale : AH + H2O = A- + H3O+
De façon générale, on admet que dans une réaction acide-base, le sens de réaction favorisé est du plus fort au plus faible membre d'un couple acide-base conjugués.
Définition : pH
Le pH d'une solution est une façon de définir le potentiel de l'ion hydronium (H3O+) par la relation pH = -log[H3O+].
Les concentrations en H3O+ et en OH- d'une eau sont liées par le produit d'ionisation de l'eau de sorte que [H3O+] [OH-] = 10-14, une solution à 25°C pour laquelle [H3O+] = [OH-] est dite neutre, son pH étant égal à 7.
Définition : Les pluies acides
Bien que l'eau pure ait un pH neutre, il n'en est pas de même pour l'eau de pluie. En effet, le pH des eaux de pluies est légèrement acide, la valeur moyenne mesurée étant pH = 5,6. Intéressons nous à l'origine naturelle de l'acidité des pluies et à la façon dont les activités humaines ont contribué à l'accentuation de l'acidité des pluies.
Le dioxyde de carbone CO2(g) est un oxyde acide qui, en se dissolvant dans l'eau forme un acide carbonique, que l'on not H2CO3 ou (CO2, H2O) :
CO2(g) + H2O = H2CO3
H2CO3 + H2O = HCO3- + H3O+ ; Ka = 4,4.10-7
C'est au cours de cette première étape d'ionisation de cet acide que l'acidité des pluies apparaît.
Par temps d'orage, lors de la formation des éclairs, la réaction à haute température entre N2(g) et O2(g) de l'air favorise la formation de NO(g). Cette réaction est suivie par l'oxydation de NO(g) et NO2(g). L'acide nitrique est alors formé par la réaction :
3 NO2(g) + H2O(l) = 2 HNO3(aq) + NO(g)
Ainsi l'acide nitrique produit naturellement contribue à l'acidité des pluies. Cependant la source de HNO3 la plus importante dans l'atmosphère provient de la source artificielle de NO(g) issue des procédés de combustion à haute température qui se produisent dans les moteurs de voiture ou dans les centrales électriques. L'acide nitrique est responsable pour ¼ de l'acidité des pluies acides.
Le dioxyde de soufre est un constituant mineur parmi les gaz atmosphérique. Dans la nature il est produit au cours de la dégradation biologique et l'activité volcanique. Les sources artificielles de SO2(g) sont beaucoup plus nombreuses. Les principales sources sont l'utilisation de combustibles riches en soufre dans les centrales électrique, ainsi que l'extraction de métaux dans les minerais sulfurés. Dans l'atmosphère, SO2(g) est oxydé en SO3(g) qui est l'acide anhydre de l'acide sulfurique. L'acide sulfurique est responsable de plus de la moitié de l'acidité des pluies acides.
Les conséquences des retombées atmosphériques acides sont de plusieurs types. D'une part, les retombées acides favorisent la dégradation des monuments réalisés en matériaux sensibles au pH tels que le marbre par exemple. D'autre part, d'un point de vue écologique, la diminution du pH des eaux (douces ou marines) peut à terme entraîner la disparition des crustacées, dont les coquilles, essentiellement composées de CaCO3, se dissoudraient dans un environnement acide. Au niveau des sols, les pluies acides provoquent une importante lixiviation des nutriments, ce qui aurait pour conséquence un appauvrissement des sols en nutriments et une croissance des plantes défavorisée.
Définition : Alcalinité et réserve alcaline
La réserve alcaline ou alcalinité correspond au calcul d'une « concentration » de charge électrique. Pour calculer la réserve alcaline d'une eau, il est nécessaire de distinguer les espèces dites « actives » et les espèces dites « inactives ». Pour cela, la représentation graphique des couples acide-base constituant une eau sous la forme logC= f(pKa) est utile. Par exemple, pour une eau de rivière dont la composition est donnée en mmol/kg dans le tableau ci-dessous :
Na+ | 0.3 | Cl- | 0.2 |
K+ | 0.05 | SO42- | 0.2 |
Ca2+ | 0.5 |
| 1 |
Mg2+ | 0.2 | H4SiO4 | 0.6 |
Les pKa des couples acide-base correspondants sont donnés Ca2+/CaOH+ = 12.7 ; Mg2+/MgOH+ = 12 ; HSO4-/SO42- = 2 ; H2CO3/HCO3- = 6.4 ; HCO3-/CO32- = 10.3 ; H4SiO4/H3SiO4- = 9.82. NaOH et KOH sont des bases fortes, leurs acides conjugués Na+ et K+ sont donc des acides dits « inactifs ». De la même façon, HCl étant un acide fort, sa base conjuguée est une base dite « inactive ». Le graphique 5b représente les espèces sous la forme logC= f(pKa). Les espèces actives sont les espèces contenues à l'intérieur de la zone délimitée par les deux droites obliques, c'es à dire H2CO3, H4SiO4, HCO3-.
L'électroneutralité de ce système s'écrit : [Na+] + [K+] + 2[Ca2+] + 2[Mg2+] + [H+] = [Cl-] + 2 [SO42-] + [HCO3-] + 2 [CO32-] + [H3SiO4-] + [OH-] ; si l'on regroupe d'un côté toutes les espèces inactives on obtient : [Na+] + [K+] + 2[Ca2+] + 2[Mg2+] - [Cl-] - 2 [SO42-] =[HCO3-] + 2 [CO32-] + [H3SiO4-] + [OH-]- [H+]. La réserve alcaline correspond à la somme des espèces inactives.
La réserve alcaline ou alcalinité représente la capacité de neutralisation des acides. Dans une eau « pure » en équilibre avec le CO2(g), elle se calcule par la relation :
Alcalinité = [HCO3-] + 2 [CO32-] + [OH-] - [H3O+]
L'alcalinité est une grandeur indépendante de la force ionique de la solution, de la température et de la pression. C'est une grandeur conservative à condition qu'aucune des espèces inactives n'intervienne dans une autre réaction chimique. Dans les eaux de pluie, la réserve alcaline est généralement négative car la composition des eaux de pluies présente un déficit de cations inactifs par rapport aux anions. Dans les eaux superficielles, la réserve alcaline est comprise entre 10-5 et 10-3 mol/kg. Dans les eaux de mer, où l'équilibre stationnaire est atteint, la réserve alcaline est constante et en moyenne égale à 2.3.10-3 mol/kg.
L'état d'équilibre acido-basique d'une eau naturelle (pour laquelle le couple carbonate est le couple acido-basique prédominant) peut être déterminé par la connaissance de la réserve alcaline et de la concentration totale en carbonates dans la solution.
Du point de vue expérimental, la détermination du titre alcalimétrique (TA) et du titre alcalimétrique complet (TAC) permet de connaître les doses en carbonates, bicarbonates et hydrates alcalins d'une eau (cf T.P).
Définition : Solubilité
Les équilibres de solubilités sont responsables de la formation ou de la dissolution des roches dans l'environnement. Le produit de solubilité correspond à la constante d'équilibre entre un solide dissout et son ion dans une solution saturée. Par exemple, le gypse (CaSO4, 2H2O) est un minerai calcique peu soluble dans l'eau, les eaux souterraines traversant des terrains riches en gypse contiennent du sulfate de calcium dissout. Les eaux riches en sulfate de calcium ne peuvent pas être utilisées dans les circuits de refroidissement des centrales nucléaires car le sulfate de calcium pourrait précipiter et boucher les circuits. L'équilibre de solubilité peut être écrit de la façon suivante :
CaSO4(s) = Ca2+ + SO42-
La constante d'équilibre de cette réaction s'écrit Ksp = [Ca2+] [SO42-]
Dans le cas d'une solution saturée de CaSO4, c'est à dire lorsque la réaction est à l'équilibre, le principe de Le Châtelier s'applique. En effet, si une quantité d'ions Ca2+ ou SO42- est ajoutée dans la solution, l'équilibre est perturbé, alors la réaction se produira dans le sens de la précipitation de CaSO4.
Ainsi nous pouvons définir les critères pour la précipitation. Considérons Q, le produit ionique des ions Ca2+ et SO42-. Lorsque :
- Q = Ksp, alors nous sommes dans les conditions d'une solution à l'équilibre avec le minéral
- Q > Ksp la solution est sur-saturée vis-à-vis du minéral, il y a précipitation de CaSO4 jusqu'à ce que la solution ait une composition respectant la condition d'équilibre.
- Q < Ksp, la solution est sous-saturée vis-à-vis du minéral. En absence de solide la solution est stable, et en présence de solide il y aura dissolution du solide jusqu'à que la composition de la solution respecte la condition d'équilibre.
Le concept de produit de solubilité (Ksp) est applicable dans le cas de solutés légèrement solubles comme le gypse, il l'est également dans le cas de composés ioniques fortement solubles comme NaCl par exemple. Cependant pour ce type de composés, il est préférable de raisonner sur les activités ioniques plutôt que sur les concentrations. En effet, dans des solutions ioniques de fortes concentrations, les activités ne sont plus assimilables à des concentrations...
Une autre limitation du concept Ksp est rencontrée lorsqu'un autre ion que les ions intervenant dans l'équilibre est présent dans le milieu. Dans ces conditions, la solubilité augmente, cet effet est appelé effet de sel. Une conséquence de cet effet de sel est que la valeur de Ksp, basée sur la molarité des espèces, dépendra de l'environnement ionique.
Dans l'expression de Ksp on considère que les solutés dissous sont présent sous forme de cations et d'anions indépendants. Cependant, le soluté peut ne pas être complètement ionique, certains solutés peuvent être présents sous forme moléculaire, ou sous forme de paire d'ions. Le degré de formation de paire d'ions augmente lorsque l'attraction électrostatique entre les anions et les cations augmente, ce qui est d'autant plus favorisé que les cations et les anions sont porteurs de charges multiples.
Dans une solution complexe, des équilibres simultanés peuvent se produire i.e. des équilibres acide-base et/ou la formation de complexes, ce qui peut entraîner des erreurs dans les calculs basés sur Ksp.
Un des principaux minéraux présent dans l'environnement issu de la précipitation est la calcite, par la réaction : Ca2+ + 2 HCO3- = CaCO3(s) + H2O(l) + CO2(g)
La calcite est également formée par l'intermédiaire de processus biologiques permettant la formation des coquilles de certains organismes marins. Ces organismes extraient les ions Ca2+ et HCO3- de l'eau de mer et les concentrent dans leurs cellules. L'organisme sécrète ainsi les cristaux de CaCO3 pour former leur carapace. La formation des carapaces est donc dépendante de la concentration en HCO3- et en Ca2+. HCO3- est un composé amphotère, il est la base conjuguée du couple CO2,H2O/HCO3- et l'acide du couple HCO3-/CO32-. Les pKa de ces couples sont respectivement 6.4 et 10.3. La zone de pH dans laquelle HCO3- prédomine est comprise entre 7.4 < pH < 9.3. Nous avons vu précédemment que l'augmentation des émissions de CO2 était responsable de la diminution du pH des eaux de surface. Une conséquence supplémentaire de ce phénomène est la diminution des épaisseurs de coquille chez les organismes marins. En effet, une diminution de pH en dessous de pH 7.4 aurait pour conséquence une forte diminution de la concentration en HCO3- dans l'eau, limitant ainsi la production de CaCO3 par les organismes marins, et menaçant leur survie.
Définition : Réactions redox
Les réactions d'oxydo-réduction dans l'environnement peuvent être d'origine biologique (activité biologique) ou chimique (réaction en milieu oxique ou anoxique). Un des éléments les plus complexe et les plus sensible aux conditions d'oxydo-reduction d'un milieu est le soufre. En effet, le soufre possède de nombreux degrés d'oxydation, entre –II et +VI. La forme la plus oxydée du soufre est constituée de sulfates (degré d'oxydation +VI) alors que la forme la plus réduite est constituée de sulfures (degré d'oxydation –II). En fonction du pH, la concentration en sulfures dissous se répartit entre les trois espèces H2S, HS-, S2-. Les sulfures associés au soufre peuvent former des polysulfures qui contribuent à la formation de sulfures de fer et à la complexation avec des métaux présents à l'état de traces dans l'environnement.
Le cycle du soufre est complexe car il comprend de nombreuses réactions intermédiaires :
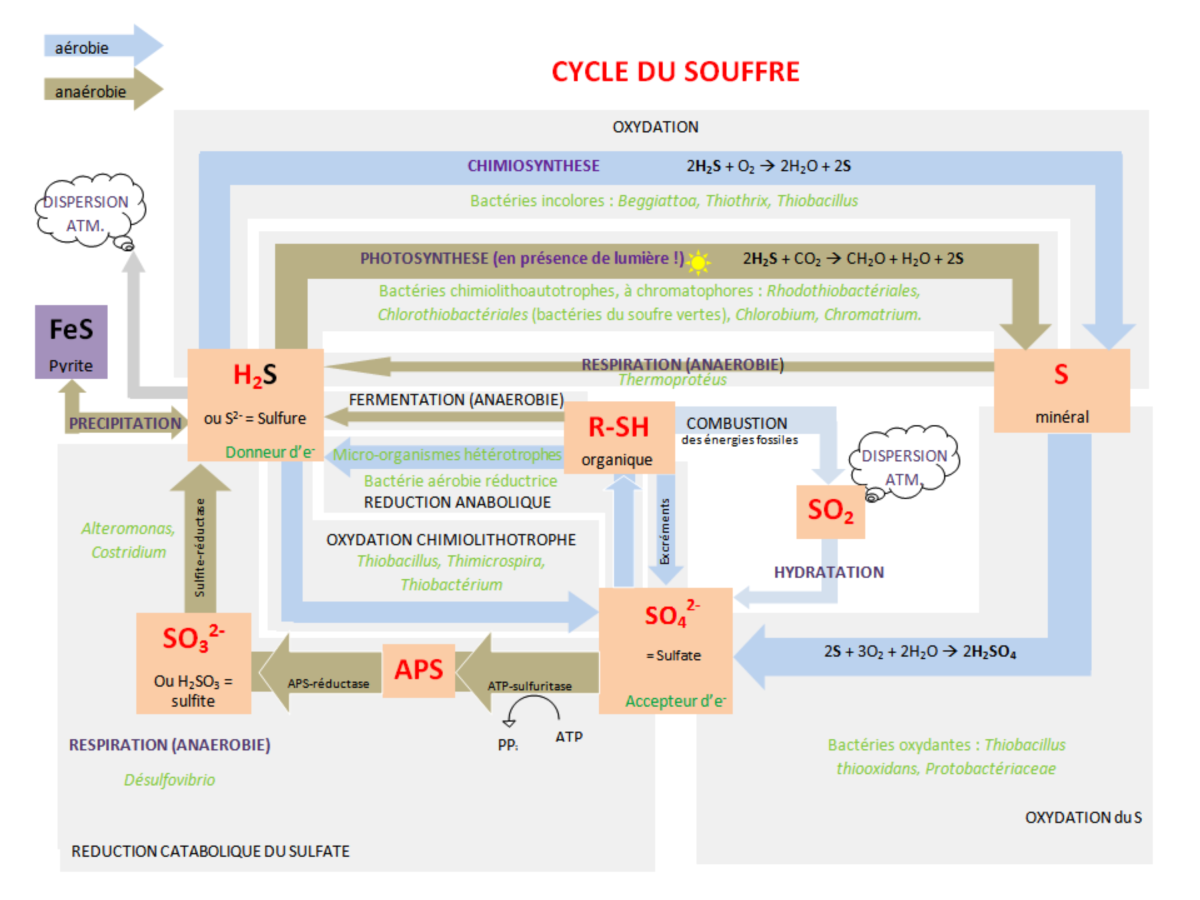
Par exemple lorsque H2S est oxydé, de nombreux composée intermédiaires tels que le soufre élémentaire des thiosulfates (S2O32-), des sulfites (SO3-) sont produits et consommés soit par respiration bactérienne, oxydation chimique ou précipitation. Les composés minéraux sulfurés formés participent activement à la séquestration des métaux traces dans le compartiment sédimentaire, c'est pour cette raison que les sédiments sont reconnus comme des « pièges » à éléments trace métalliques. Dans l'environnement, l'eau peut agir comme un agent réducteur, dans ce cas H2O est réduit en H2 :
2H+ + 2e- = H2(g) E° = 0V
ou comme un agent oxydant, dans ce cas H2O est oxydé en O2.
4H+(aq) + O2(g) + 4e- = 2H2O(l) E° = 1,23V
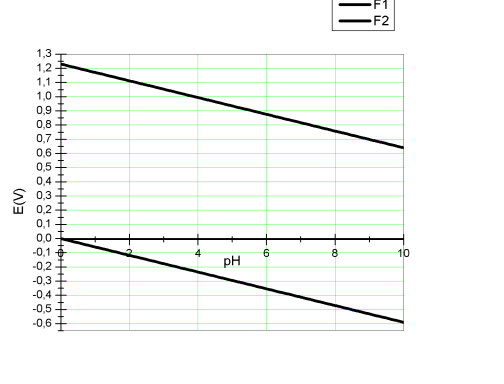
Le domaine de stabilité de l'eau correspond au domaine de potentiel et de pH pour lequel l'eau est thermodynamiquement stable. Les limites du domaine de stabilité de l'eau correspondent à l'équation E = f(pH) pour chacune des 2 demi-réactions précédemment citées. La relation de Nernst écrite pour chacune des 2 demi-réactions nous permettra d'obtenir les limites du domaine de stabilité de l'eau.
Pour l'équation 12, la relation de Nernst donne : E = E° + RT/4F ln(pO2 [H+]4), soit pour pO2 = 1bar ; E(V) = 1,23 -0.059pH.
Ainsi toute espèce dont le potentiel de réduction est supérieur à l'équation ci-dessus sera réduite par l'eau, la réaction est alors accompagnée par un dégagement de O2.
Pour l'équation 13, la relation de Nernst donne E = E° -RT/2F ln(pH2/[H+]2), soit pour pH2 = 1bar ; E(V) = -0.059pH.
Toute espèce dont le potentiel d'oxydation est inférieur à cette équation pourra réduire H+ en H2. Les couples redox thermodynamiquement stables dans l'eau sont compris entre les limites inférieures et supérieures du domaine de stabilité de l'eau. Le domaine de stabilité dans les eaux naturelles est représenté en ajoutant deux lignes verticales à pH=4 et pH=9 qui délimitent le domaine de pH qui couvre les pH des eaux naturelles.
En plus du rôle de l'eau en tant qu'agent oxydant ou réducteur, on peut s'attendre à des réactions d'oxydation entre les solutés et l'oxygène dissous dans le cas de système ouvert (c'est-à-dire lorsqu'il y a contact entre l'eau et l'air). La chimie des eaux naturelles peut être comprise par l'utilisation des diagrammes de Pourbaix. Ainsi lorsqu'une eau est en contact avec l'atmosphère, elle est saturée en oxygène et de nombreuses espèces peuvent être oxydées. Les formes les moins oxydées seront rencontrées dans des eaux profondes, où la teneur en oxygène est faible, et plus particulièrement en présence de matière organique qui agit comme un agent réducteur. Le système qui contrôle le pH des eaux naturelles est le système calco-carbonique. L'activité biologique joue également un rôle important puisque la respiration consomme l'oxygène et dégage du CO2. A l'opposé, la photosynthèse consomme le CO2 et dégage de l'O2. Ces différentes réactions vont contribuer à l'évolution du pH d'un milieu aqueux naturel. Le schéma 8 représente le domaine de prédominance des différents types de système aquatique, en fonction du potentiel E(V) et du pH du milieu.

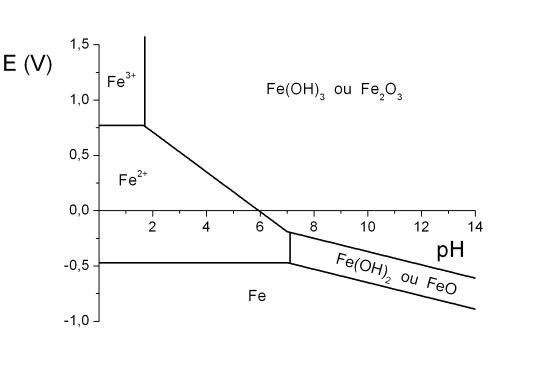
Pour mieux comprendre les réactions redox susceptibles de se produire dans l'environnement, la représentation de la prédominance d'un élément sous ses différents degrés redox en fonction du potentiel et du pH est souvent utilisée (diagramme de Pourbaix) (exemple du diagramme de Pourbaix du fer schéma 9). Ainsi il est facile, en comparant les diagrammes de Pourbaix de l'eau, le diagramme de Pourbaix d'une espèce chimique et le diagramme de prédominance des différents systèmes aquatiques (schéma 8), de connaître la forme prédominante d'une espèce chimique en fonction des conditions E-pH du milieu dans lequel elle se trouve.