Le point sur les endosomes (précoces et tardifs) et les lysosomes (primaires et secondaires)
Les endosomes forment un compartiment cellulaire issu du processus d'endocytose, caractérisé par l'invagination de la membrane plasmique et la formation de vésicules (mécanisme très proche de la formation des vésicules bourgeonnantes des REr et Golgi). Ces vésicules fusionnent entre elles pour former un compartiment de forme irrégulière appelé endosome précoce. L'endosome précoce se transforme en endosome tardif (ou corps multivésiculaire) caractérisé par de nombreuses invaginations et vésicules internes. L'endosome tardif se transforme en lysosome (ou encore lysosome secondaire). Enfin, une fois le contenu dégradé le lysosome gagne l'état de corps résiduel (ce sujet est abordé en détail à la fin de cette ressource dans le chapitre "Les endosomes").
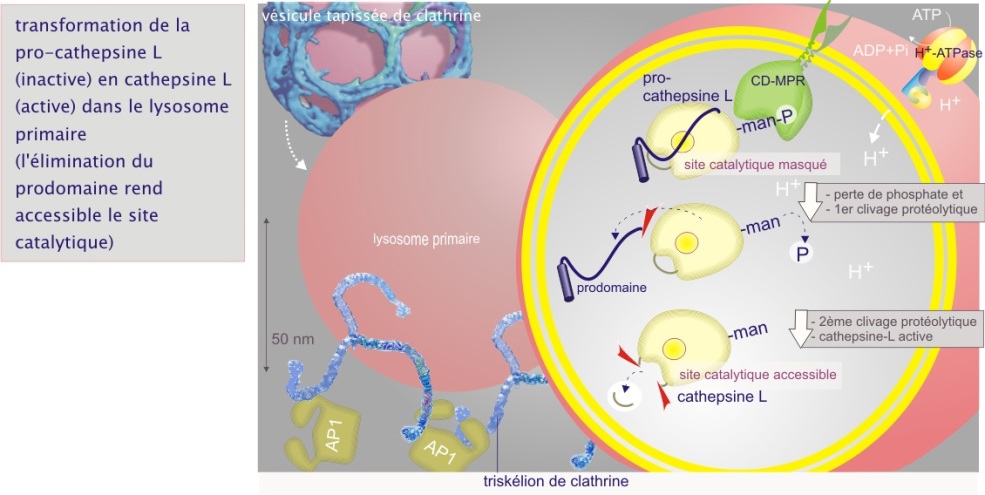
Les transformations successives surviennent en raison de la fusion des vésicules riches en hydrolases (lysosomes primaires) provenant du réseau trans-golgien avec les endosomes précoces et tardifs. Les transformations sont accompagnées par une acidification progressive du contenu endosomal due à l'intervention d'une pompe H+–ATPase type V (vésiculaire). Dans le contexte de la maturation des hydrolases, l'acidification a deux effets : 1/ découplage de la charge protéique de son récepteur et 2/ protéolyse partielle (par une cystéine protéase) de la pro-hydrolase. La pro-cathepsine L (murine) possède trois sites de clivage : après le résidu 144, pour enlever le pro-peptide, après les résidus 288 et 290 pour former une chaîne lourde et une chaîne légère (). Le mannose perd enfin son phosphate et l'enzyme est alors prête à agir (voir figure 3).
pour plus de détail sur les sites de clivage de la cathepsine L (murine).
La membrane du lysosome primaire contient un lipide original : l'acide lyso-biphosphatidique. Elle contient également de nombreuses protéines transmembranaires, les pompes et transporteurs, mais, de loin, les protéines les plus abondantes sont les Lamps (lysosome-associated membrane protein) et la Limp (lysosome integral membrane protein), toutes deux fortement N–glycosylées. Elles sont résistantes aux activités des hydrolases. La présence de motifs GY ou IL permet à ces protéines de lier directement, au niveau du réseau trans golgien, les protéines du manteau AP–1A (ou AP3).
Complément : Partez en excursion : les hydrolases du lysosome primaire
Ici, nous traiterons des différentes hydrolases présentes dans le lysosome primaire et des pathologies associées à leur absence ou à leurs anomalies. Nous montrerons aussi comment expérimentalement l'α–galactosidase, en modifiant la composition de la chaîne de glucides, convertit l'antigène du groupe sanguin B en O.
A ce jour, on connaît environ 40 hydrolases différentes (hydrolases lysosomales) : protéases, lipases, phospholipases, glycosidases et nucléases, elles sont toutes apportées par des vésicules issues du Golgi.
Protéases
Parmi les protéases, les cathepsines dominent et sont majoritairement responsables de la dégradation des protéines, comme nous l'avons déjà évoqué dans la ressource « ribosomes et protéasomes : la synthèse et la dégradation des protéines ».
Glycosidases
Les glycosidases aussi sont nombreuses : arylsulfatases, \(\alpha\)–fucosidase, \(\beta\)–galactosidase, \(\alpha\) et \(\beta\)–glucosidase, \(\beta\)–glucuronidase, \(\beta\)–hexosaminidase, \(\alpha\)–iduronidase, \(\alpha\)–mannosidase, \(\beta\)–mannosidase, n–acétyl–\(\alpha\)–glucosaminidase, parmi d'autres. Elles débarrassent les protéines, lipides et glycogène, de leurs sucres. Le manque d'activité de certaines de ces enzymes est à l'origine de maladies dites de surcharge (lysosomal storage diseases). En effet, les lysosomes accumulent des débris cellulaires incomplètement détruits, en particulier des glycolipides et du glycogène. Les conséquences en sont désastreuses.

Par exemple, dans la maladie de Fabry, les sphingoglycolipides s'accumulent à la suite du manque d'–galactosidase, enzyme qui doit nécessairement détacher le galactose terminal, pour que les autres glycosidases puissent dégrader la chaîne glucidique (voir figure 30). Cette maladie récessive, décrite en 1898 par J. Fabry, est liée au chromosome–X. Elle se caractérise par une douleur chronique, des opacités oculaires, un dysfonctionnement hépatique et rénal, des lésions cutanées et vasculaires et une déficience cardiaque. De nombreuses mutations ponctuelles sont à l'origine de cette maladie ; pour voir les sites mutés de l'enzyme,.
Fabry, J. (1898). Ein Beitrag Zur Kenntnis der Purpura Haemorrhagica nodularis (Purpura papulosa hemorrhagica Habrae). Arch. Dermatol. Syph. 43, 187
L'administration intraveineuse d'\(\alpha\)–galactosidase humaine recombinante, sous le nom commercial de Replagal ou Fabrazyme, restaure la fonction enzymatique des patients. C'est un des premiers exemples de thérapie correctrice réussie par administration d'une protéine recombinante produite par la technologie dite de génie génétique.
Un autre exemple de maladie de surcharge lysosomale est la maladie de Pompe, décrite en 1932 par J. Pompe. Elle est la conséquence de la perte d'activité de l'\(\alpha\)–glucosidase et se traduit par une accumulation de glycogène dans le lysosome qui, à son tour, entraîne en l'absence de traitement, l'atrophie musculaire et la mort du patient vers l'âge de 2 ans. La thérapie actuelle est l'administration de l'enzyme recombinante (Myozyme™).
Bien qu'on ne sache pas exactement comment le glycogène entre dans le lysosome, on soupçonne l'intervention d'un processus d'autophagie. La taille des lysosomes devient alors impressionnante, de l'ordre de 5 (normalement environ 0,4 \(\mu m\)). La figure 31 montre une coupe longitudinale d'un muscle d'une souris dépourvue du gène de l'\(\alpha\)–glucosidase.

pour plus de détail sur la séquence d'alpha-1,4-glucosidase humaine.
Les groupes sanguins A, B, AB et O
L'\(\alpha\)–galactosidase recombinante a retenu l'attention pour sa capacité potentielle d'interconversion des antigènes de groupes sanguins. En effet, les groupes sanguins humains, A, B, AB et O, sont dus à la présence de chaînes oligosaccharidiques spécifiques à la surface des hématies. Un individu du groupe A ne peut recevoir du sang du groupe B, car ses anticorps feraient précipiter les « hématies B ». De même un individu du groupe B ne peut recevoir de « sang A ». Un individu du groupe O ne pourra recevoir ni « sang A » ni « sang B » (et à plus forte raison du « sang AB »). Les membres des groupes A, B et AB peuvent recevoir le « sang O ». Par action de l'\(\alpha\)–galactosidase recombinante, qui enlève le galactose terminal du groupe B, le sang B peut être transformé en sang O (voir figure 6). Ceci peut avoir un intérêt dans la gestion des stocks de sang destiné à la transfusion.

Lipases
On trouve également des enzymes d'altération des lipides: lipase acide, sphingomyélinase , palmitoyl–protéine thioestérase, parmi d'autres. Les mutants de ces enzymes sont aussi à l'origine de maladies de surcharge telles que la lipofuscinose (maladie neurodégénérative caractérisée par une accumulation de lipopigments autofluorescents, conséquence de l'absence de palmitoyl–protéine thioestérase), la maladie de Niemann-Pick (manque de sphingomyélinase) et la maladie de Wolman (manque de lipase acide), pour lesquelles il n'existe pas encore de thérapie.
ADNase et ARNase
Désoxyribonucléase II (ADNase), ectonucléoside triphosphate diphosphohydrolase–4 (UDPase) et ARNase. Aucune maladie connue n'a été reliée au manque d'une de ces enzymes.