Les caractéristiques de l'interaction récepteur-ligand
La liaison du ligand (endogène) à son récepteur est dans la plupart des cas non covalente. Pour les petits ligands, hormones et neurotransmetteurs par exemple, l'interaction est déterminée par des liaisons très localisées de nature électrostatique (longue et courte distance, attractive ou répulsive) et du type forces de « van der Waals » (courte distance, toujours attractive). Pour les grands ligands il s'ajoute une attraction hydrophobe qui concerne des surfaces très étendues. L'interaction est réversible, le ligand s'associe à son récepteur puis, après un certain temps, s'en sépare.
Lorsque l'on étudie la liaison ligand-récepteur dans un espace temporel donné (quelques secondes), et qu'on mesure la durée de l'association, une durée faible caractérise un ligand à faible affinité (pour son récepteur), alors qu'une longue durée caractérise un ligand à forte affinité. Pour occuper le maximum de récepteurs pendant un certain temps, un ligand de faible affinité doit donc être présent en forte concentration (de l'ordre du nanomolaire (nM)). (les ligands se remplacent et gardent les récepteurs occupés).
Au contraire, une faible concentration suffit (de l'ordre du picomolaire (pM)) pour un ligand de forte affinité (voir figure 6).
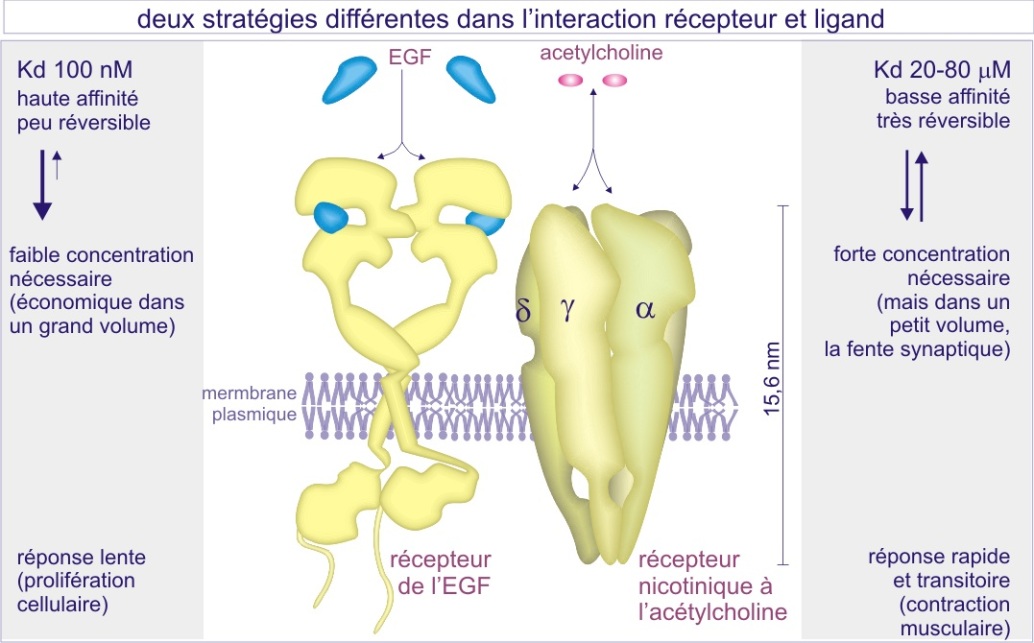
L'avantage de la faible affinité est la réversibilité rapide (de l'ordre de la milliseconde) de l'interaction alors que l'avantage de la forte affinité est une économie dans la production du ligand lui-même. L'acétylcholine, dont le récepteur est un canal sodium/potassium, est un exemple d'un ligand de faible affinité (nM) permettant des contractions musculaires transitoires et rapides. L'EGF est un exemple de ligand à haute affinité (pM) permettant l'induction de la prolifération cellulaire à faibles concentrations. Nous reviendrons sur ce sujet dans les paragraphes suivants.
Les récepteurs de surface sont en général au nombre de 10.000 copies par cellule. Ceci est nécessaire pour assurer un signal même en cas de faible concentration du ligand et donc de faible pourcentage de récepteurs occupés.
Si du point de vue pharmacologique, les récepteurs et les seconds messagers vous intéressent vous trouverez ci-dessous une série de liens avec des articles qui listent les récepteurs classiques, leurs noms, leurs ligands, leur localisation tissulaire, leurs agonistes et antagonistes. Ces articles ont été publiés sous l'entête « Receptor Listing » dans la revue British Journal of Pharmacology, volume 144, Issue Supplement 1 (Mars 2005). Cependant, vous ne trouverez rien sur les récepteurs tyrosine kinase, sérine/thréonine kinase ou sur les récepteurs de cytokines car les modalités d'interaction ligand récepteur n'ont pas encore été explorées par l'intermédiaire de drogues agonistes ou antagonistes.
Pour en savoir plus, consultez les documents PDF suivants :
« Guide to receptors introduction Alexander [pdf] » (61 Ko).
« Key to tables Alexander [pdf] » (36 Ko).
« Ion-channelsAlexander [pdf] » (272 Ko).
« Ligand-gated ion channels Alexander [pdf] » (182 Ko).
« GPCR 7TM Alexander [pdf] » (514 Ko).
« Catalytic receptors Alexander [pdf] » (79 Ko).
« Nuclear receptors Alexander [pdf] » (92 Ko).
« Second messenger metabolizing enzymes Alexander [pdf] » (115 Ko).
« Transmitter transporters Alexander [pdf] » (116 Ko).
Complément : Excursion 2 : Quantification de l'interaction récepteur-ligand ; Kd ou Ka.
Liaison du ligand à son récepteur
Une hormone typique induit des effets physiologiques dès la concentration de 10-10M, ce qui signifie 10-10 x 6 x 1023 (nombre d'Avogadro) = 6 x 1013 molécules par litre. Pour avoir une idée du nombre de molécules qui rencontreront une cellule cible, considérons qu'une cellule d'un diamètre de 12 µm occupe un volume de 10-12 litre. Ce volume contient 60 molécules d'hormone susceptible de contacter la cellule par fixation non-covalente bien que spécifique. Pour que l'effet de l'hormone soit significatif, il faut que la cellule « capture » le maximum de molécules d'hormones présent en si faible quantité. Ceci s'effectue par l'intermédiaire d'un récepteur cellulaire de forte affinité. Il est important de savoir que la liaison de l'hormone à son récepteur est réversible.
On peut donc écrire l'interaction hormone (H) récepteur (R) comme suit :
![]()
A l'équilibre, l'état ou le nombre de récepteurs occupés est constant, le taux de réaction d'association (forward reaction) est équivalent au taux de réaction de dissociation (backward reaction) et selon la loi d'action de masse
![]()
où kon est la constante d'association et koff la constante de dissociation. Leurs unites sont mol-1 s-1 et s-1, respectivement. [H] et [R] représentent la concentration d'hormone libre et la concentration de récepteurs libres. Dans le cas de haute affinité (kon est forte et koff faible), la configuration dominante serait HR, l'équilibre est déplacé vers la droite ou, en d'autres termes, la constante d'équilibre « K » est grande.
![]()
Les unités de K sont l mol-1 (c-a-d concentration réciproque). On peut aussi inverser l'expression ci-dessus et obtenir, plutôt qu'une constante d'équilibre d'association (K), une constante de dissociation (K D ) qui a l'avantage de justifier d'unités de concentration simples (mol l-1) :
![]()
Cette constante est souvent utilisée pour exprimer l'affinité d'un ligand pour son récepteur, indication importante pour l'emploi thérapeutique des agonistes et antagonistes : un faible K D se traduit par une forte affinité du récepteur pour son ligand, ce qui signifie également qu'une très faible concentration du ligand (agoniste ou antagoniste) sera efficace.
Hétérogénéité de la liaison
On pourrait imaginer que tous les récepteurs sont égaux en termes d'effets qu'ils engendrent en réponse à la fixation de leur unique ligand. Mais c'est loin d'être le cas. L'hétérogénéité des sites de liaison peut être causée par l'existence d'isoformes de récepteurs, avec des affinités différentes, ou par des récepteurs de même isotype (classe) soumis à des oscillations entre l'état de haute affinité et l'état de faible affinité (transition conformationnelle). Ces oscillations sont souvent induites par des modifications intracellulaires (fixation d'une autre protéine ou phosphorylation).
Mesure de l'affinité de liaison
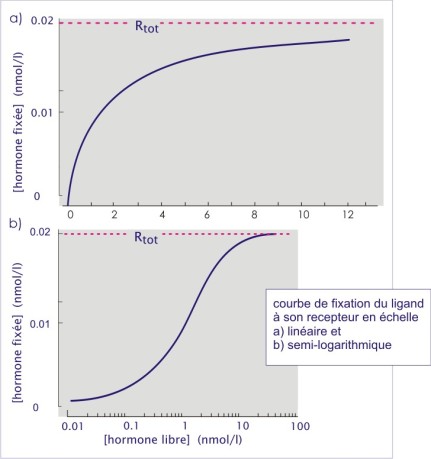
Pour évaluer les paramètres de liaison, on expose les cellules ou leur membrane à l'hormone radio marquée. Lorsque l'équilibre est atteint, l'hormone libre est séparée de l'hormone liée, généralement par centrifugation ou filtration. Le surnageant ou le filtrat contient le matériel non lié et le précipité ou le filtre, contient les cellules ou les membranes ayant lié l'hormone marquée. Après lavage du filtre ou du précipité la quantité de radioactivité est mesurée dans les deux fractions. La spécificité est déterminée par la même expérience avec excès d'hormone non radio-marquée (hormone froide). L'affinité est déterminée en appliquant des doses croissantes d'hormone radio marquée (jusqu'à saturation). L'équation de la courbe de liaison peut s'écrire comme suit :
![]()
Rtot représente la concentration totale des récepteurs dans la préparation. Si on substitue [R] dans l'équation (ci-dessus) on obtient l'équation d'équilibre de liaison concernant un seul site :
![]()
Si l'on représente [HR] en fonction de [H], on obtient une hyperbole. Le plus souvent, l'échelle de grandeur de la concentration de H étant très large, on adopte une représentation logarithmique. Sur le graphique, l'extrapolation pour des concentrations infinies d'hormone, permet d'obtenir la concentration totale des récepteurs [Rtot ] dans le système expérimental étudié. Avec ce graphe on peut également calculer la constante de dissociation (K D ) (indice de l'affinité du récepteur pour son ligand).
KD et EC50 : liaison du récepteur et ses conséquences fonctionnelles
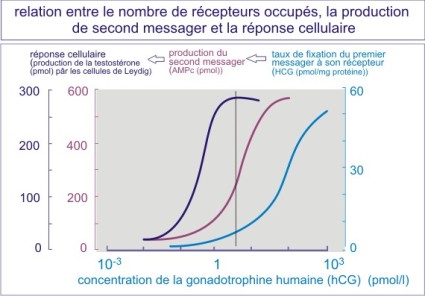
Le K D renseigne sur l'interaction ligand - récepteur mais pas sur la réponse physiologique due à la liaison du ligand. Un autre indicateur est donc nécessaire : il s'agit de l'EC50 qui représente la concentration d'hormone qui induit la moitié de la réponse physiologique maximale (concentration efficace (EC)) qui donne lieu à une réponse de 50%). La figure E06 illustre la contraction du muscle lisse en réponse à l'adrénaline . On peut voir qu'un très faible nombre de récepteurs occupés induit déjà un maximum de réponse contractile. La relation entre le nombre de récepteurs occupés et l'intensité de la réponse est le plus souvent non-linéaire.
On peut également évaluer la réponse à un autre niveau, tel que celui des seconds messagers. Un exemple est donné par l'action de la human Chorionic Gonadotrophin (hCG) sur la production de testostérone par les cellules de Leydig du testicule. On peut voir qu'un très faible nombre de récepteurs à l'hCG occupés induit déjà une forte production d'AMPc et qu'une faible concentration d'AMPc se traduit déjà par une forte production de testostérone. Il s'agit là d'un exemple typique d'amplification du signal qui rend le système très sensible au ligand.
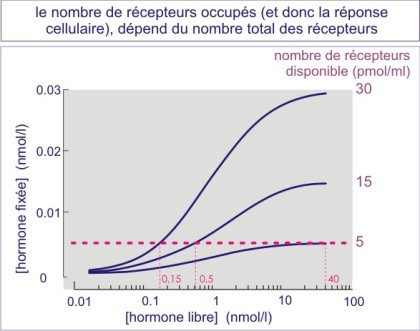
Un nombre élevé de récepteurs rend la cellule plus sensible au ligand
Le nombre de récepteurs occupés par cellule à une concentration donnée du ligand, dépend du nombre total des récepteurs exposés en surface (voir figure E07). Les cellules qui expriment des nombres élevés de récepteurs sont donc plus sensibles au ligand que les cellules qui en expriment moins. On peut voir ici la justification de l'excès de récepteurs qu'on trouve dans la plupart des cellules. On peut aussi comprendre pourquoi une surexpression aberrante peut induire un comportement anormal de la cellule (sans que la concentration du ligand change). Par exemple la surexpression du récepteur d'EGF (ErbB2) par des cellules épithéliales mammaires, entraîne leur prolifération excessive et les rend invasives.
« Down regulation » des récepteurs
Comme nous l'avons montré dans la ressource 07 « acheminement des récepteurs à travers le REr et le Golgi », les récepteurs occupés par leur ligand sont souvent internalisés et transportés dans un compartiment intracellulaire appelé (dans certain cas pour y être détruits, dans d'autres cas pour y être recyclés après avoir été dissociés de leur ligand). L'ensemble du processus conduit au phénomène connu sous le nom de « down regulation » des récepteurs qui permet à la cellule de diminuer le nombre de ses récepteurs exposés et donc la sensibilité à son ligand (rétrocontrôle négatif). Une forte présence de ligand pendant un certain temps, peut éventuellement conduire à une désensibilisation complète de la cellule.
L'intervention sur la concentration en seconds messager modifie la sensibilité au ligand
On peut modifier l'EC50, sans changer l'affinité du ligand pour son récepteur, par manipulation des enzymes impliquées dans la production/élimination des seconds messagers intracellulaires. Un bon exemple est donné par la théophylline (l'un des alcaloïdes du thé), médicament utilisé pour traiter les symptômes de l'asthme bronchique et certaines maladies respiratoires (telles que « chronic obstructive pulmonary disease » (COPD)). La théophylline est un inhibiteur de la phosphodiestérase, enzyme responsable de la conversion d'AMPc en 5'-AMP. Elle augmente la réponse bronchodilatatrice en agissant en aval du récepteur b 2 de la noradrénaline. C 'est en effet, par l'intermédiaire de son récepteur b 2, que la noradrénaline induit la production d'AMPc par stimulation de l'adénylate cyclase. Normalement, l'AMPc est rapidement éliminé par la phosphodiestérase. L 'inhibition de cette dernière par la théophylline augmente considérablement l'AMPc disponible et donc l'effet bronchodilatateur de la noradrénaline (diminution de l'EC50), qui améliore la ventilation.
Pour plus d'information nous suggérons un excellent article par D. Jenkinson dans « Textbook of Receptor Pharmacology » Eds Foreman, Johansen, Gibbs. 3ème édition (2010), CRC Press, Royaume Uni.